
Abstract
This work illustrated the mechanism contributing to the process of Phosphatidylinostiol 3-kinase (PI3K)/protein kinase B (PKB)/mammalian target of rapamycin (mTOR) signaling pathway, which has been demonstrated to play an important role in virus-induced apoptosis, which contributes to the Viral Myocarditis (VMC) pathogeneses. We examined the expression of Bax, Bim, caspase-3, caspase-9, and viral replication after Coxsackievirus B3 (CVB3) infection using the mTOR inhibitor and PI3K inhibitor pretreated HeLa cells, respectively. Apoptosis in different groups was determined by flow cytometry. Bax, Bim, caspase-9, and caspase-3 were examined by semiquantitative polymerase chain reaction (PCR) and Western blot analysis. The expression of CVB3 mRNA and viral capsid protein VP1 were analyzed by semiquantitative PCR and Western blot analysis distinctively. We found that rapamycin and LY294002 promote CVB3-induced cytopathic effect (CPE) and apoptosis. CVB3 replication in host cells is mediated in mRNA and protein expression by rapamycin and LY294002. Moreover, comparing with controls, at 12 and 24 h of postinfection (p.i.), Bim and Bax expression increased in cells after treated with rapamycin or LY294002, which also stimulates the activation of procaspase-9, and the CVB3-induced caspase-3 self-cleavage. However, in the meantime, the mRNA expression of caspase-9 and caspase-3 did not have an obvious change. In summary, our results demonstrated that the mTOR-signaling pathway plays an important role in CVB3-induced CPE and apoptosis, which is indispensable in VMC, via regulating Bim, Bax, caspase-9, caspase-3, and viral replication. Our findings may provide a new perspective and a deeper understanding of the mechanism of CVB3-induced apoptosis which, in turn, may help with the development of new therapy for the CVB3 infection.
Introduction
Viral Myocarditis (VMC) is one of the common acquired heart diseases in children and presents a spectrum of symptoms. In severe cases, malignant arrhythmia, cardiac failure, cardiogenic shock, or even sudden death may occur, and the persistent ones may develop to the dilated cardiomyopathy. Especially, it has a characteristic that, the younger the age the higher the morbidity. In many viruses causing VMC, Coxsackievirus B is one of the most common pathogens, of which, Coxsackievirus B3 (CVB3), a small, nonenveloped, positive-strand RNA Enterovirus in the Picornaviridae family, is thought to be the main one because of its strongest myocardial affinity. VMC mainly performs in the inflammatory lesions of the myocardial caused by the viral infection and it is well known to be an immune-mediated disease (Coronado et al., 2012; Liao et al., 2012). However, it has reported recently that the apoptosis of myocardial cells also plays an indispensable role in pathogenesis of the VMC (Frustaci, 2010; Liao et al., 2012), of which, the mechanism is not very clear yet.
Bax and Bim are well known to be the members of BH-only proteins, the pro-apoptosis in the BcL-2 family. Bim is expressed in many tumor cells and is vital for the maintenance of homeostasis in the hematopoietic system as a new identity—the barrier against autoimmune disease (Hughes et al., 2006). It has been long reported that Bax can be directly activated by Bim to trigger cytochrome c release and therefore promote the caspase activation and cell death, which is a major core of the intrinsic apoptosis pathway at the mitochondria (Lindsten et al., 2000). Caspase, a family of cysteine protease, is the common mediator of apoptosis induced by various stimuli, of which, caspase-3 contributes to the apoptosis execution, while caspase-9 participates in the apoptosis origination. In the present study, we offer new insights into the potential role of the mTOR signal pathway playing in the CVB3-induced cytopathic effect (CPE) and apoptosis through regulating of the pro-apoptosis factors.
Materials and Methods
Cell culture and virus propagation
HeLa cells (HeLa S3) were obtained from the Institute of Oncology, Central South University and preserved in the liquid nitrogen pipe. Cells were grown in the Dulbecco's modified Eagle's medium (DMEM, Gibco, Life Technologies, Inc.) containing 10% heat-inactivated fetal bovine serum (FBS; Gibco, Life Technologies, Inc.) at 37°C in a humidified incubator with 5% CO2. Coxsackievirus B3 (CVB3) Nancy strain was obtained from the Shanghai Jiao Tong University School of Medicine, propagated in HeLa cells, and stored at −80°C in our laboratory. The titer of virus was routinely determined before each experiment.
Virus infection
HeLa cells were infected at multiplicity of infection of 10 with CVB3 or with the DMEM containing 2% FBS for sham. After 1 h of infection, cells were washed with phosphate-buffered saline (PBS) and replenished with a fresh DMEM containing 2% FBS. For inhibitor experiments, HeLa cells were pretreated with the mTORC1 inhibitor (Rapamycin; Cell signaling Technology) or PI3K inhibitor (LY294002; Cell signaling Technology) or Dimethyl Sulfoxide (DMSO; Amresco) for control or the DMEM containing 10% FBS for sham, 30 min later, cells were washed with PBS and replaced with a medium containing fresh inhibitors or the DMEM containing 2% FBS for sham.
MTT assay
HeLa cells were seeded in 96-well plates and divided into sham, DMSO, rapamycin, and LY294002 groups. After being washed twice with PBS, cells were treated with rapamycin in 1, 10, 50, 100 nM or LY294002 in 5, 10, 25, 50, 100 μM for experiment groups, 0.1% DMSO for control, and the DMEM containing 10% FBS for sham. Cells were grown at 37°C in a humidified incubator with 5% CO2 for 24 h. Then, the medium was replaced by the serum-free DMEM, and 20 μL of the MTT solution was added. After further growth at 37°C in a humidified incubator with 5% CO2 for 4 h, the supernatant was discarded carefully, and then 150 μL DMSO was added to each well. After vibrated for 10 min, the optical density of each well was measured on the enzyme-linked immunosorbent detector at 560 nm wave length.
Cell apoptosis analysis
Apoptosis was determined by fluorescence-activated cell sorting (FACS) analysis of cells stained with Annexin-V FITC and propidium iodide (PI, Promega) by a flow cytometer. At each time point, cells were harvested using 0.25% EDTA-Trypsin (Gibco, Life Technologies, Inc). After centrifugation, cell pellets were washed twice with cold PBS, and then the cells pellets were incubated with Annexin V-FITC and propidium iodide to achieved double staining, according to the manufacturer's instructions. The mixture was incubated in the dark for 15 min at room temperature. Afterward, 400 μL of a 1× binding buffer was added to each tube and cells were immediately analyzed by FACSCalibur flow cytometry (Becton Dickinson).
Semiquantitative PCR
HeLa cells either untreated or treated with different inhibitors were harvested at each time point and first had mRNA extracted following the method of RNA Extracted Kit (Omega). Next, mRNA was reverse transcribed into cDNA following the RevertAid™ First Strand cDNA Synthesis Kit (Thermo). At last, PCR amplification was done, while referring to this article (Liu et al., 2003). The primer sequences and PCR conditions are on the tables below (Tables 1 and 2).
This table contains the sequences of primers used in the process of semiquantitative PCR.
PCR, polymerase chain reaction.
This table contains the PCR conditions in the process of semiquantitative PCR.
Western blot analysis
HeLa cells either untreated or treated with different inhibitors were washed twice with ice-cold PBS, and then kept on ice for 10 min. Then, 80 μL of the lysis buffer (Beyotime Institute of Biotechnology) containing 0.1% phenylmethylsulfony (PMSF; Cwbio) was added in each well. Afterward, cell lysates were collected by scraping and centrifuged at 12000 rpm for 15 min at 4°C, and then the precipitate was discarded. The protein concentration was determined by the Enhanced BCA protein Assay Kit (Beyotime Institute of Biotechnology). Thirty micrograms of extracted protein was fractionated on sodium dodecyl sulfate-10% to 12% polyacrylamide gels, electrophoretically transferred to 0.45 μm PVDF membranes (Millipore Corporation), and blocked with deionized water containing 10% nonfat dry milk and bovine serum albumin for 1 h. Afterward, the membrane was incubated with the primary antibody (monoclonal anti-enterovirus antibody (Dako Co.), monoclonal anti-bim antibody (Cell signaling technology), polyclonal anti-bax antibodies (Cell signaling technology), monoclonal anti-caspase-9 antibody (Cell signaling technology), polyclonal anti-caspase-3 antibodies (Cell signaling technology), and monoclonal anti-β-actin antibody (Proteintech Group, Inc.)) at 4°C overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Beijing Boisynthesis Biotechnology Co.). At last, protein expression was detected by enhanced chemiluminescence (Pierce Professional Resources).
Statistical analysis
Two-way analysis of variance with multiple comparisons and paired Student's t-tests were performed. Data are presented as the mean±standard error (SE). The p value of <0.05 was considered significant.
Results
Both rapamycin and LY294002 inhibit cell viability in a concentration-dependent manner
PI3K/Akt/mTOR is one of the most important signaling pathways in cell growth and reproduction. To test the effect, we determined the cell viability by MTT assay with mTORC1-specific inhibitor rapamycin and PI3K-specific inhibitor LY294002. HeLa cells were treated with rapamycin or LY294002 for 24 h at different concentrations. We found that both inhibitors can suppress the cell viability. Besides, the higher the concentration, the stronger the suppression effect (Fig. 1).
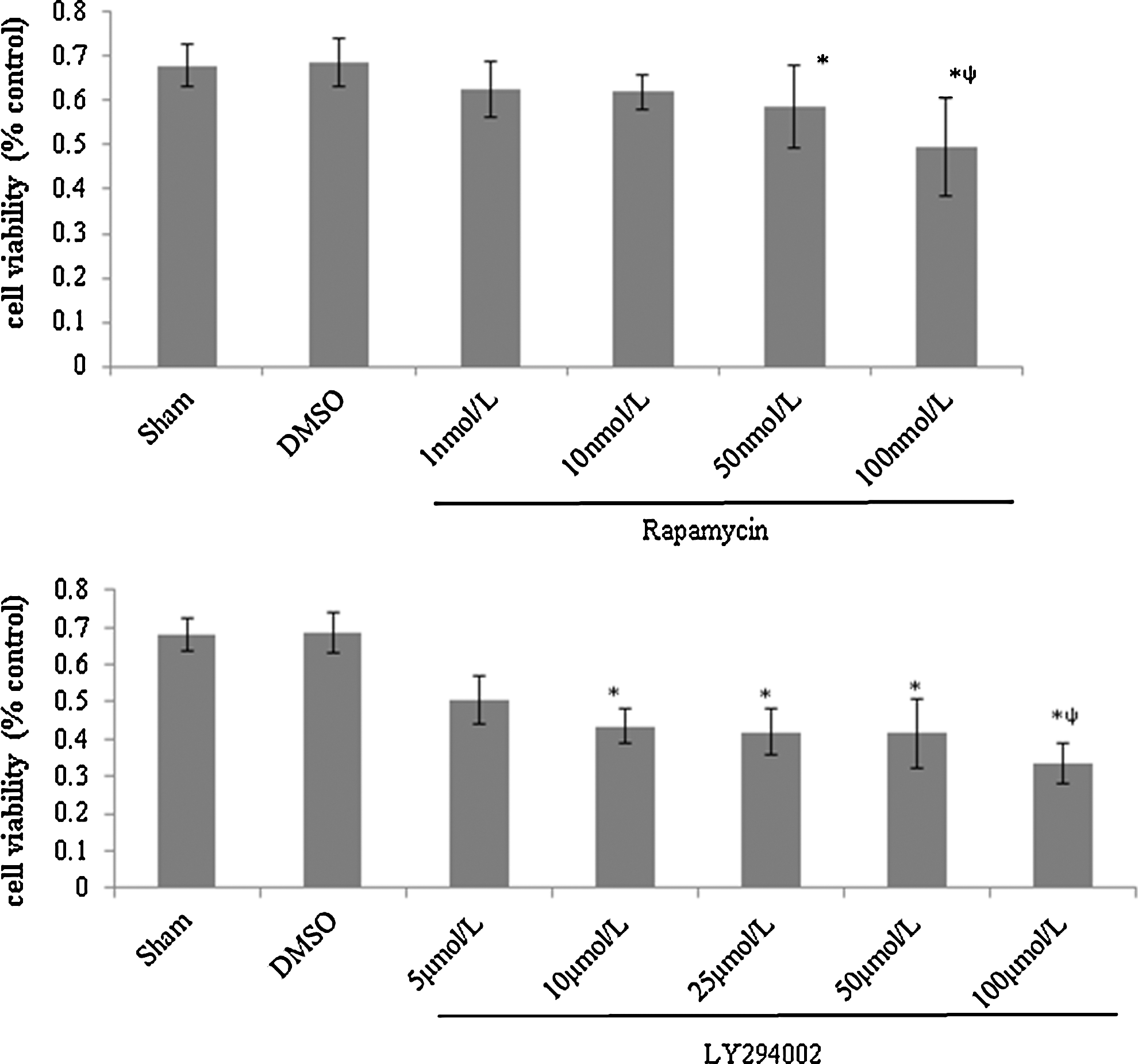
Both rapamycin and LY294002 inhibit cell viability in a concentration-dependent manner. HeLa cells were divided into sham, rapamycin, LY294002, and Dimethyl Sulfoxide (DMSO) groups, treated with rapamycin in 1, 10, 50, 100 nM or LY294002 in 5, 10, 25, 50, 100 μM for experiment groups, 0.1% DMSO for control, and DMEM containing 10% FBS for sham, cell viability was determined by MTT assay. Compared with the DMSO group, rapamycin in 50, 100 nM and LY294002 in 10, 25, 50, 100 μM were significant differences (*p<0.05). Comparisons among different concentrations in the rapamycin or LY294002 group, rapamycin in 100 nM and LY294002 in 100 μM were significant differences (ψ p<0.05).
Rapamycin and LY294002 promote CVB3-induced CPE and apoptosis
Recent studies have shown that CVB3-induced apoptosis is involved in the VMC pathogenesis, of which, CPE is a typical feature, characterized by degenerative changes in cell morphology, including cell shrinkage, cell rounding, and eventually cell detachment. Our previous research had showed that the mTOR signal pathway participated the viral replication (Chen et al., 2007), and to further test it, we examined the role it plays in the CVB3-induced apoptosis by flow cytometry. HeLa cells, pretreated with rapamycin, LY294002, or DMSO, were infected with CVB3 and harvested at 3, 6, 12, and 24 h p.i. Consistent with our expected results, the apoptosis of cells with rapamycin and LY294002 were obviously increased and CVB3-induced CPE had progressed in these groups (Fig. 2).
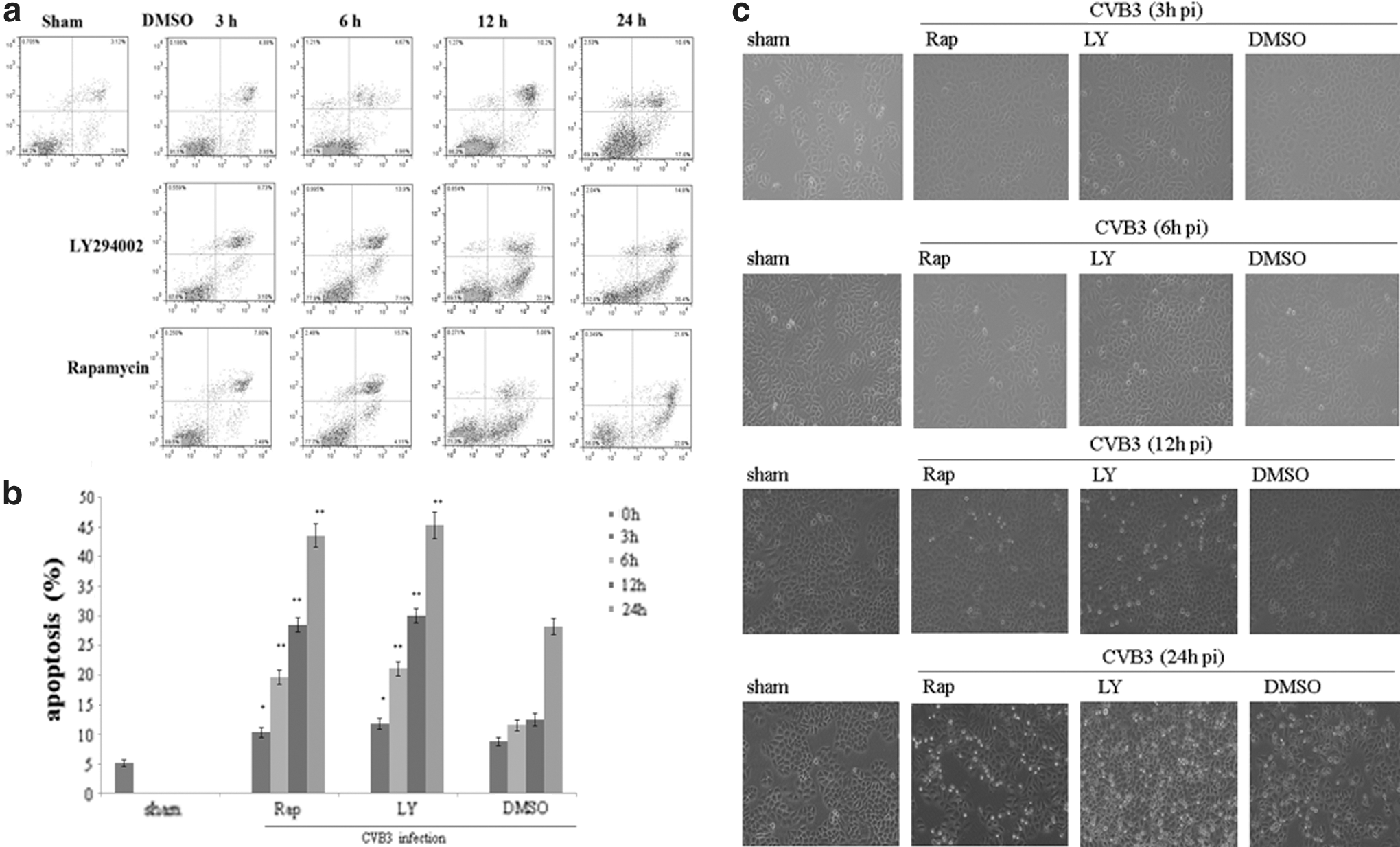
Rapamycin and LY294002 promote Coxsackievirus B3 (CVB3)-induced cytopathic effect (CPE) and apoptosis. HeLa cells were divided into sham, rapamycin, LY294002, and DMSO groups, treated with rapamycin in 10 nM or LY294002 in 25 μM or 0.1% DMSO, cells were infected with CVB3. Collected at 3, 6, 12, and 24 h p.i., apoptosis was determined by flow cytometry immediately after dyed by Annexin V and P.I. (mean±SE, n=3)
LY294002 stimulates viral mRNA synthesis and blocks viral protein VP1 expression promoted by rapamycin
It has been demonstrated that CVB3 infection can lead to the phosphorylation of PKB/Akt though a PI3K-dependent mechanism, which stimulates mTOR and its downstream factors contributing to host defense against CVB3 infection in return (Liu et al., 2012). To further characterize the change of viral replication when this pathway was blocked, HeLa cells pretreated with rapamycin (10 nM), LY294002 (25 μM) were infected with CVB3, and we examined mRNA and viral capsid protein VP1 by semiquantitative PCR and Western blot analysis, respectively. Cell lysates were collected at 3, 6, 12, and 24 h p.i. We found that LY294002 stimulated the CVB3 mRNA expression, but blocked the VP1 expression, while rapamycin mainly stimulated the VP1 expression, especially at 24 h p.i., but had no effect on the mRNA expression. After 3 h of infection, the VP1 expression was still zero, which was an unexpected result for us (Fig. 3 and Table 3A, B). Owing to the different mediated ways of CVB3 mRNA and VP1 expression and the affirmatory role of CVB3 in cardiomyocyte apoptosis, it is conceivable to consider that the PI3K/Akt/mTOR signal pathway indeed plays a role in the CVB3-induced apoptosis, in addition, the regulation of CVB3 replication via different ways is one of its putative mechanisms.
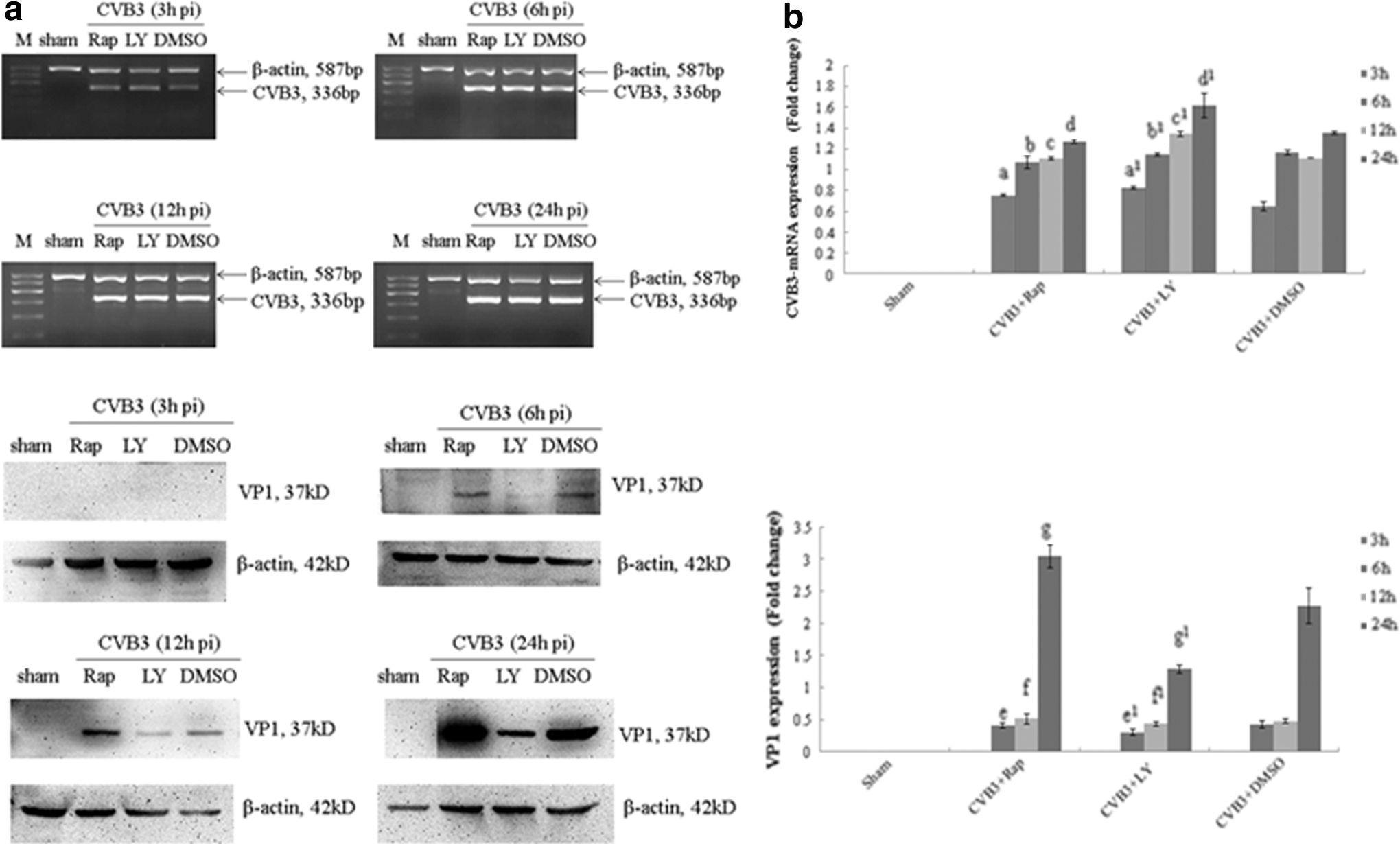
LY294002 stimulates viral mRNA synthesis and blocks viral protein VP1 expression promoted by rapamycin. HeLa cells, pretreated with rapamycin (10 nM), LY294002 (25 μM), or DMSO (0.1%) were infected with CVB3.Cell lysates were collected at 3, 6, 12, and 24 h postinfection (p.i.). CVB3 mRNA and VP1 activity were determined by semiquantitative PCR and Western blot analysis
CVB3, Coxsackievirus B3; DMSO, dimethyl sulfoxide.
Rapamycin and LY294002 block the ability of CVB3 to prevent Bim activation
Previous results have shown that the blockage of mTORC1 or PI3K can promote the CVB3-induced apoptosis. Being a pro-apoptosis factor, Bim is activated via the JNK and p38 kinase pathways and CHOP during ER stress (Lei and Davis, 2003; Puthalakath et al., 2007; Brem et al., 2012), and has been reported to directly link ER stress to mitochondrial apoptosis in cells expressing mSOD1 (Soo et al., 2012). To figure out the relationship between Bim and the mTOR signal pathway, we determined the Bax mRNA and protein expression by semiquantitative PCR and Western blot analysis using rapamycin or LY294002 pretreated cells. We found that the change of the Bim activity was dynamic. After CVB3 infection, LY294002 could promote the protein expression, which was decreased in accordance with the infected time increasing, and stimulated the mRNA expression in 3, 12, and 24 h p.i. significantly. Meanwhile, rapamycin stimulated the protein expression at 12 h p.i. (Fig. 4 and Table 4A, B).
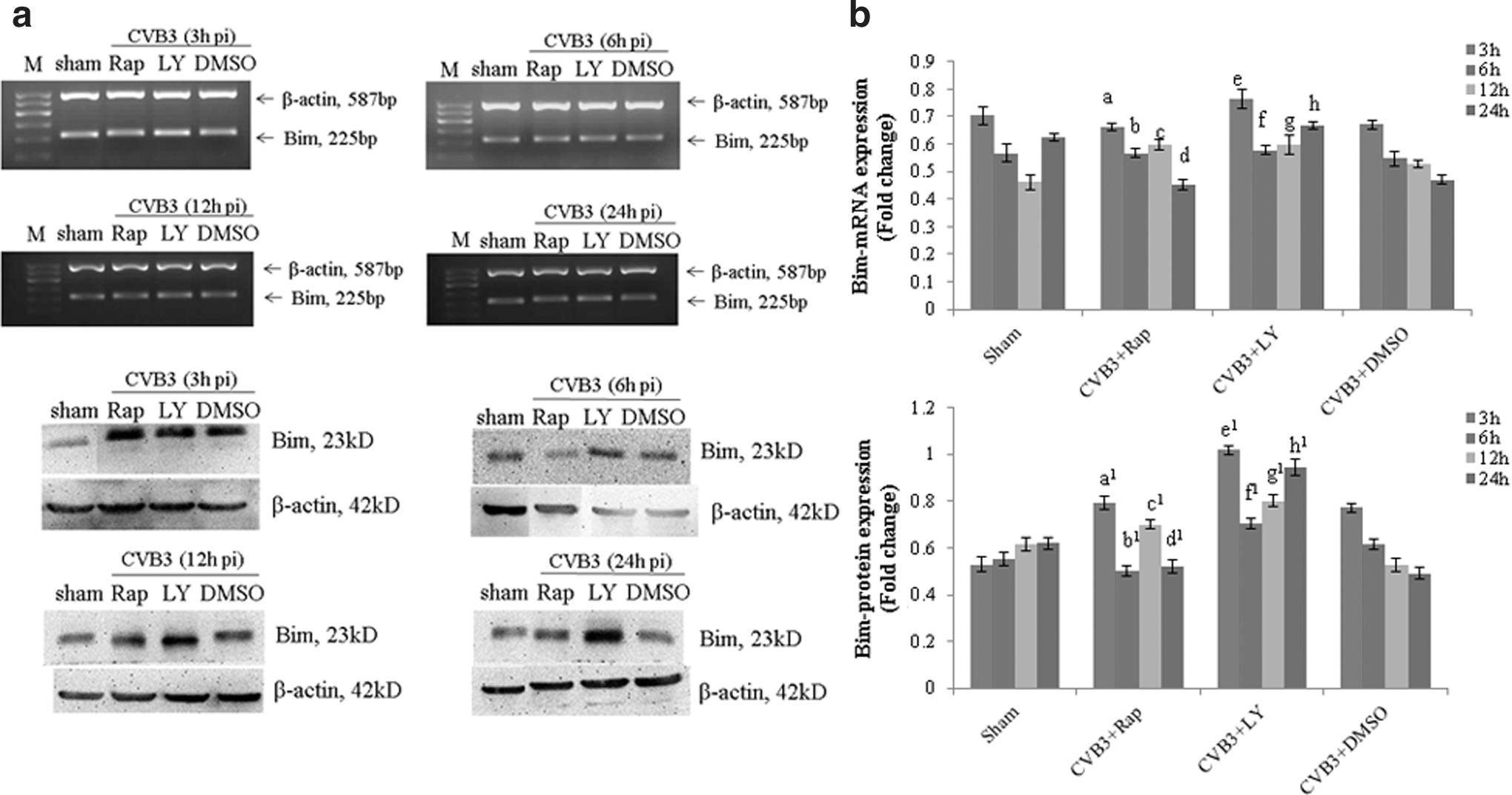
Rapamycin and LY294002 block the ability of CVB3 to prevent Bim activation. HeLa cells, pretreated with rapamycin (10 nM), LY294002 (25 μM), or DMSO (0.1%), were infected with CVB3. Cell lysates were collected at 3, 6, 12, and 24 h p.i. The Bim activity was determined by semiquantitative PCR and Western blot analysis
Rapamycin and LY294002 stimulate the CVB3-induced Bax activation
Bax has been reported to be the major core of the intrinsic apoptosis pathway at the mitochondria, which is directly mediated by Bim activation. To investigate whether Bax expression can be mediated by the mTOR signal pathway, which is closely related to the Bax expression, Bax mRNA and protein were analyzed by semiquantitative PCR and Western blot analysis using rapamycin or LY294002 pretreated cells HeLa cells pretreated with rapamycin (10 nM), LY294002 (25 μM), or DMSO (0.1%) were infected with CVB3. Cell lysates were collected at 3, 6, 12, and 24 h p.i. To our surprise, the change of Bax mRNA and protein expression was opposite. The protein expression was increasing with the viral infected time increasing, which was simulated by rapamycin and LY294002, while the mRNA expression was decreased in the case of LY294002 only (Fig. 5 and Table 5A, B). Bax activation promotes the cytochrome c release to initiate the caspase cascade, which provides us a way to track.
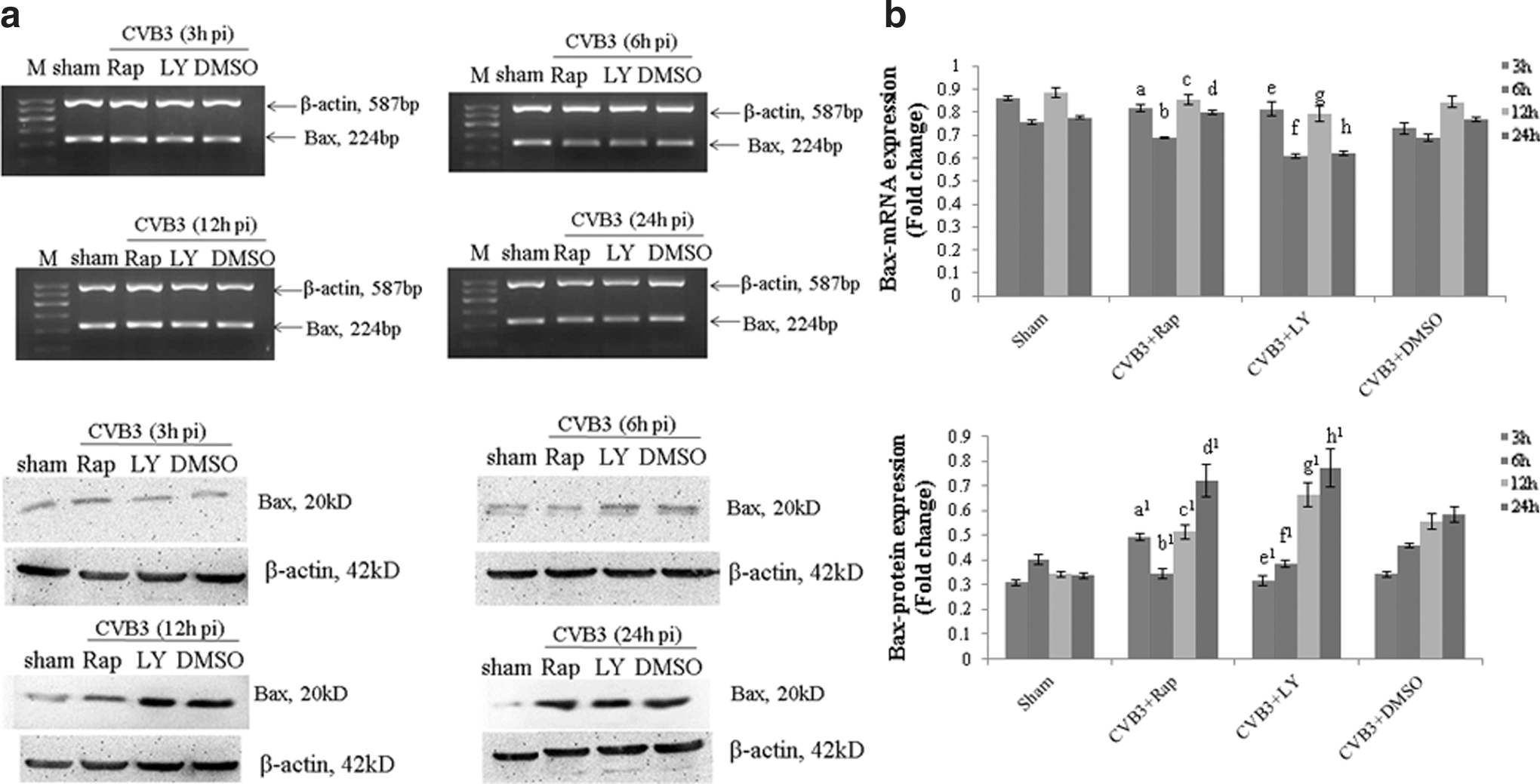
CVB3-induced Bax mRNA and protein expression change can be dynamically mediated by rapamycin and LY294002. HeLa cells, pretreated with rapamycin (10 nM), LY294002 (25 μM), or DMSO (0.1%), were infected with CVB3. Cell lysates were collected at 3, 6, 12, and 24 h p.i. The Bax activity was determined by semiquantitative PCR and Western blot analysis
Rapamycin and LY294002 stimulate CVB3-induced procaspase-9 activation to initiate a caspase cascade
Caspase, a cysteine protease family, is one of important effector molecules during apoptosis. Upon apoptotic stimulation, cytochrome c released from mitochondria associates with the procaspase-9/Apaf 1, which processes procaspase-9 self-cleavage. Cleaved caspase-9 further processes other caspase members, including caspase-3, leading to apoptosis. To test the role of the mTOR signal pathway in the activation of the caspase family during apoptosis, we first determined the caspase-9 activation by semiquantitative PCR and Western blot analysis using the monoclonal antibody. HeLa cells were pretreated with rapamycin, LY294002, and cell lysates were collected at 3, 6, 12, and 24 h p.i. Both rapamycin and LY294002 promoted the procaspase-9 activation at 12 and 24 h p.i., while the mRNA expression did not change obviously. To our surprise, the mRNA expression in the LY294002 group at 6 h p.i. was decreased, but caspase-9 was increased significantly. Together with the change of cleaved caspase-9, except the increased level at 24 h p.i., our results revealed an approximate decrease in the level of cleaved caspase-9 concurrently with an increase in procaspase-9 protein (Fig. 6 and Table 6A–C).
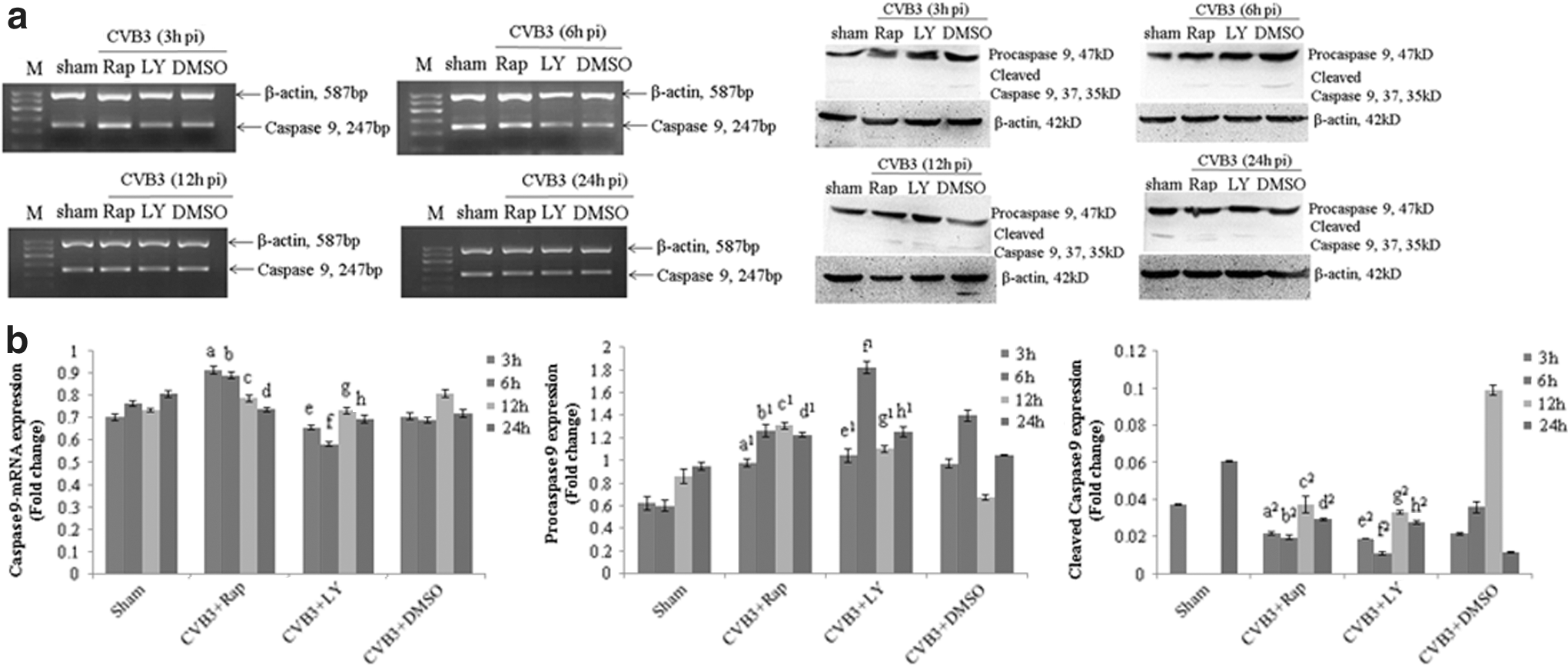
Rapamycin and LY294002 stimulates CVB3-induced procaspase-9 activation to initiate a caspase cascade. HeLa cells, pretreated with rapamycin (10 nM), LY294002 (25 μM), or DMSO (0.1%), were infected with CVB3. Cell lysates were collected at 3, 6, 12, and 24 h p.i. The caspase-9 activity was determined by semiquantitative PCR and Western blot analysis
Rapamycin and LY294002 stimulate CVB3-induced caspase-3 self-cleavage
Caspase-3 is a critical executioner of apoptosis, as it is either partially or totally responsible for the proteolytic cleavage of many key proteins such as the nuclear enzyme poly polymerase (PARP). In this research, caspase-3 mRNA and protein expressions were analyzed with semiquantitative PCR and Western blot analysis using rapamycin or LY294002 pretreated cells. HeLa cells were pretreated with rapamycin, LY294002, and cell lysates were collected at 3, 6, 12, and 24 h p.i. Both rapamycin and LY294002 could not change the mRNA expression. However, the regulation of caspase-3 protein expression was dynamical. At an early stage after CVB3 infection, caspase-3 was stimulated, while being followed by an increase in cleaved caspase-3 concurrently with the decreased level of caspase-3 protein (Fig. 7 and Table 7A–C).
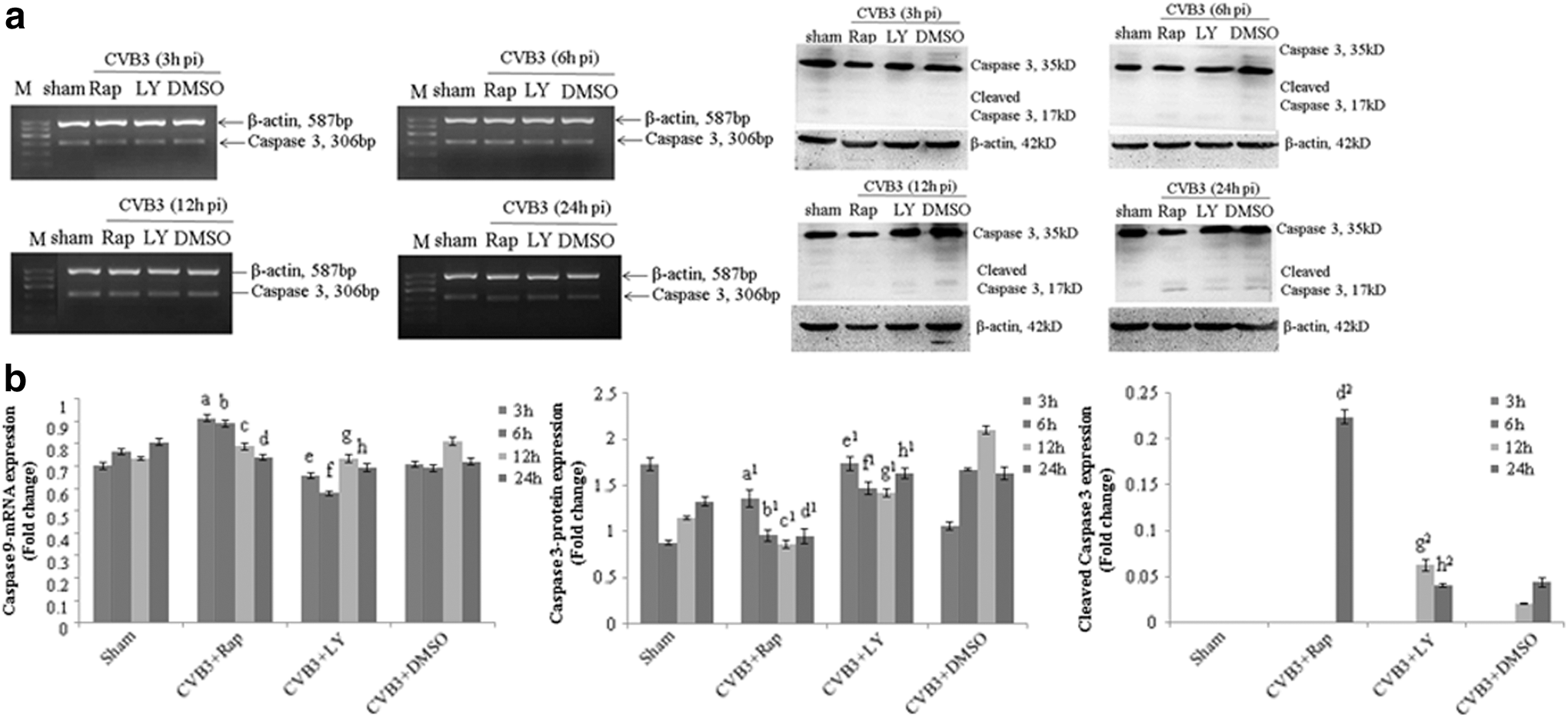
Rapamycin and LY294002 stimulate CVB3-induced caspase-3 self-cleavage. HeLa cells, pretreated with rapamycin (10 nM), LY294002 (25 μM), or DMSO (0.1%), were infected with CVB3. Cell lysates were collected at 3, 6, 12, and 24 h p.i. Caspase-3 activity was determined by semiquantitative PCR and Western blot analysis
Discussion
Apoptosis and the mTOR-signaling pathway have been demonstrated to be involved in many diseases among which, tumorigenesis is highly correlated and has been reported recently to be indispensable in VMC pathogenesis. In the present study, we have shown that both the mTOR inhibitor rapamycin and PI3K inhibitor LY294002 can promote CVB3-induced apoptosis and CPE. Inhibition of mTOR stimulates the CVB3 mRNA expression at an early time after viral infection, but later in the VP1 expression. Meanwhile, the stimulation of CVB3 mRNA expression by inhibition of PI3K stays longer after viral infection, at the same time, it blocks the VP1 expression. Together with our discoveries that the expressions of pro-apoptosis factors in Bcl-2 and caspase families change dynamically with the inhibition time of rapamycin and LY294002 after CVB3 infection, our data demonstrate that the PI3K and mTOR-signaling pathway participate in the CVB3-induced VMC through mediating the pro-apoptosis factor Bim, Bax, caspase-9, caspase-3, and the viral replication. Additionally, our findings raise a potential mechanism that inhibition of the mTOR-signaling pathway may activate some feedback signaling pathways, such as the activation of Akt at Ser473 playing a role in the regulation of apoptosis.
In recent years, mTOR has attracted increasing attention in the world as an important regulator of cell cycle and protein synthesis, which is a critical component in many signaling pathways, such as the PI3K/Akt signal, regulating cell growth via accepting exogenous growth factor and insulin stimulation, feeling the energy change, nutritional status, and so on. mTOR has two downstream signal molecules—the eIF4E-binding protein 1 (4E-BP1) and p70S6 kinase (S6K), and when mTOR is active, it phosphorylates both of them. The phosphorylation of 4EBP1, directly activated by mTORC1 stimulation, is a major point of control in cap dependence by regulating the function of the eIF4F translation initiation complex, binding to the 5′ cap of an mRNA, which is the first step in the initiation of cap-dependent translation. CVB3 infection activates the PI3K/Akt signal pathway on both Ser-473 and Thr-308 (Esfandiarei et al., 2004). mTORC2 can be activated by PI3K directly and phosphorylates Akt at Ser-473, which together with phosphorylation at T308 results in the complete activation of Akt (Nawroth et al., 2011). Therefore, mTORC1 is fully activated, and then phosphorylates 4EBP1, maintaining cap-dependent translation. It has been reported that mTORC2 is the upstream signal molecular of mTORC1, which phosphorylates Akt at Ser-473 (Moore et al., 2011; Nawroth et al., 2011), and together with another report that has presented that activation of mTORC1, mediated by PI3K, Akt, and the inhibitory tuberous sclerosis complex 1/2 (TSC1-TSC2), initiates a negative feedback loop that ultimately inhibits PI3K (Dalle et al., 2012). We speculate that the continued fully activation of the mTORC1 signal may lead to the feedback inhibition of mTORC2, thereby the phosphorylation of Akt is blocked, and then the activation of mTORC1 is inhibited.
Rapamycin and LY294002 are the specific inhibitors of mTORC1 and PI3K, respectively. After using rapamycin or LY294002 pretreated CVB3-infected HeLa cells, apoptosis and CPE were becoming more serious as the time of infection increased. Meanwhile, CVB3 replication in these inhibitor pretreated cells was also affected which, however, was not our expectation. During CVB3 infection, it hijacks the host cells to favor its own replication (Hemida et al., 2013). It has been reported that the phosphorylation of Akt on Ser-473 is only mediated by mTORC2 (Tripathy and Jump, 2013). Therefore, unlike the direct inhibition of mTORC1 by rapamycin, LY294002 specifically inhibits PI3K, which blocks phosphorylation of Akt at Thr-307 only, but CVB3 infection stimulates Akt at both Ser-473 and Thr-307, and the activation of mTORC1 is partly inhibited by LY294002. On account of the reactivation of Akt due to the rapamycin-induced mTORC2 activity (Hresko and Mueckler, 2005; Ramakrishnan et al., 2012), although nutrients were gradually depleted with the infection time and mTORC1 was further inhibited, Akt phosphorylation was increased to the contrary, which was an event upstream of IRE-JNK signaling and consequent apoptosis (Kato et al., 2012). Therefore, the inhibition of mTORC1 was blocked, and the cap-dependent translation was promoted in turn, which may contribute to the increased VP1 expression in cells treated with rapamycin. Together with its effect on viral mRNA expression, we suggest that increasing the viral protein instead of mRNA expression is one of the mechanisms that rapamycin promotes CVB3-induced CPE and apoptosis.
In cells treated with LY294002, the expression of CVB3 mRNA increased compared with the control, while the protein expression was not keeping pace. It has been reported that the Mek/Erk pathway and the PI3K/Akt pathway cross inhibit each other and because of this cross inhibition, inhibitors of either pathway lead to upregulation of the other pathway (Hoeflich et al., 2009). It has been reported earlier that inhibition of the Extracellular Signal-Regulated Kinase (ERK) signal pathway reduces the CVB3 replication (Luo et al., 2002). Therefore, ERK-mediated downstream activation of the PI3K/Akt pathway results in resistance of Akt inhibition in host cells (Leisner et al., 2012; Ramakrishnan et al., 2012), which was utilized by CVB3 to accelerate its mRNA synthesis. Nevertheless, at 24 h p.i., mRNA activation was higher than the control, which can promote the translation, but the VP1 expression was lower. As previously mentioned, partly inhibiting the activation of mTORC1 by LY294002 may lead to the feedback inhibition of Akt, which may block the activation of mTORC1 in turn and next inhibit the cap-dependent mRNA translation. Hence, the high expression of CVB3 mRNA was not contributed to the VP1 synthesis in cells treated with LY294002. Taken together, stimulating the mRNA rather than VP1 activation is one of the mechanisms that LY294002 promotes the CVB3-induced CPE and apoptosis.
Bim and Bax, as the important members of the Bcl-2 family, have been long reported to do their best to promote the apoptosis. Recently, however, another study demonstrating that there is a Bax/Bak independent intrinsic apoptosis pathway emerging from a cross talk between the Endoplasmic reticulum (ER) and the mitochondria (Zamorano et al., 2012) and Bim probably acts as a prosurvival factor in cancer cells (Gogada et al., 2013) making the previous conclusion controversial. The caspase family is an important executer of apoptosis, in which, caspase-9/3 have long been proven to be the proapoptosis factors, and caspase 8 has recently been reported to have a little relationship with viral-induced apoptosis (Imao and Nagata, 2013). ER stress, occurring as an early event that leads to mitochondrially mediated apoptosis through CHOP upregulation and subsequent Bim activation, has been implicated in various cell types, in which, Bim is essential and indispensable (Ghosh et al., 2012; Shinde et al., 2012; Soo et al., 2012). In accordance with that, showing that Bim activation is decreased at 12 and 24 h p.i., our results have confirmed that Bim is activated at the early period of apoptosis. However, in this study, when treated with rapamycin or LY294002, Bim protein expression increased at 12 and 24 h p.i., of which, inhibition of PI3K led to this increase being more significant. Bim released and translocating to Bcl-2 resulted in Bax-dependent apoptosis, through which playing its proapoptotic role. Moreover, it has been reported that Bax is directly activated by Bim activation, through which the downstream factors are stimulated to participate in the apoptosis (Cartron et al., 2012). In healthy cells, Bax is either located in the cytosol or loosely associated to the mitochondria and endoplasmic reticulum (Kudo et al., 2012). In this study, Bax activation was stimulated after CVB3 infection, which was further stimulated after using rapamycin or LY294002. CVB3 infection stimulates the PI3K/Akt signal pathway to promote the growth of the host cells, which is conducive to the viral replication. Inhibition of mTORC1 or PI3K prolongs the time of Bim activation, and further activates the Bax expression to promote the process of apoptosis, which is consistent with the study that PI3K-dependent Bax/Bak activation promotes the platelets apoptosis (Zhang et al., 2013). Activated Bax contributes to form channels on the mitochondrial membrane leading to cytochrome c release (Willis et al., 2007), which can lead to caspase-9, the initiator in the caspase family during apoptosis, self-cleave. Cleaved caspase-9 further processes other caspase members, initiating a caspase cascade, including caspase-3. Some earlier reports have shown that in addition to self-cleavage, procaspase-9 can also be cleaved in vivo by caspase 3 at Asp 330, which serves as positive feedback to amplify the apoptotic signal in the caspase activation pathway (Zou et al., 2003; Denault et al., 2007). In our study, similar to the report that the activated PI3K/Akt pathway inactivates caspase-9 and offers resistance against dexamethasone-induced apoptosis (Zong et al., 2009), the procaspase-9 expression increased at an early stage and cleaved caspase-9 was activated at 24 h p.i., which was further promoted with mTORC1 or PI3K inhibitor caspase-3 expression. However, it was different in cells treated with rapamycin or LY294002. The procaspase-3 protein expression increased at an early stage after CVB3 infection with rapamycin or LY294002 treated and decreased as the infection time prolonged, following the increased level of cleaved caspase-3, demonstrating that this protein is processed to further promote apoptosis when PI3K or mTORC1 was directly inhibited.
In conclusion, these findings suggest inhibition of mTORC1 and PI3K has different regulatory pathways in the CVB3-induced apoptosis. LY294002 promotes the apoptosis via upregulating the CVB3 mRNA expression and Bim, Bax, caspase-9, caspase-3 protein activation, while different from LY294002, rapamycin upregulates the viral protein synthesis instead of mRNA. Looking at everything as a whole, our study demonstrates that the PI3K and mTOR signal pathways participate in the CVB3-induced apoptosis through mediating the proapoptotic factors in Bcl-2 and caspase families, which provide new ideas and evidences for us to do further research about VMC.
Footnotes
Acknowledgments
This subject was funded by the Fundamental Research Funds for the Central Universities of Central South University and National Natural Science Foundation (NO. 30973233).
We appreciate that the Center Laboratory at the Third Xiangya Hospital of the Central South University provided us with the experimental equipment and technical guidance necessary to complete our work.
Disclosure Statement
No competing financial interests exist.