
Abstract
Fipronil (FP) is a phenylpyrazole pesticide developed by the transnational company Rhône-Poulenc Agro in 1987. Data on the genotoxicity and toxicity of FP are rather inadequate. In this study, we aimed to evaluate the potential genotoxic activity of FP using the single-cell microgel electrophoresis or comet assay, sister chromatid exchanges (SCEs), and micronuclei (MN) in human peripheral blood lymphocytes. In addition, the cytokinesis block proliferation index (CBPI) and proliferation index (PRI) were measured for cytotoxicity. In this study, three different doses of FP were used (0.7, 0.3, 0.1 μg/mL). Mitomycin C (2 μg/mL) and hydrogen peroxide were used as positive controls for SCE MN test systems, and comet assay, respectively. FP induced a statistically significant increase in the MN and SCE frequency and DNA damage in a dose-dependent manner in human peripheral blood lymphocytes (p<0.01, p<0.05, for 0.7 and 0.3 μg/mL, respectively) compared with a negative control. There is no significant difference between 0.1 μg/mL and the negative control for MN frequency, but there is significant difference between all the doses of FP and negative control for SCE frequency, mitotic index, CBPI, and PRI values (p<0.01). Using the alkaline comet assay, we showed that all the doses of the FP induced DNA damage in human peripheral blood lymphocytes in vitro (p<0.05).
Introduction
T
The demonstrated harmful effects of many pesticides, such as organochlorines, organophosphorus, and carbamates, and the imperative to use pesticides that are safe for nontarget organisms, whenever possible, have led to the use of different group of pesticides as alternatives. In fact, these pesticides are known to possess high activity against a broad spectrum of insect pests (both adults and larvae), low acute toxicity in mammals, and lack of persistence in the environment (Papadopoulou-Markidou, 1983; Zerba, 1988; Vijveberg and Van den Bercken, 1990). Pesticides may also affect long-term human health, due to the persistence and bioaccumulation of toxic pollutants in the food chain (Alves, 1990; Houk, 1992; World Health Organization, 1992).
Fipronil (FP) is a phenylpyrazole pesticide developed by Rhône-Poulenc Agro in 1987. FP is available in a range of formulations (from attractive baits to sprays) in veterinary products in the United States. FP is highly efficient in control of pests, including those resistant to pyrethroid, organophosphate, and carbamate insecticides (Kidd and James, 1991; Higgins et al., 2001).
FP is an extremely active molecule and a potent disruptor of the central nervous system of invertebrates and shows effect by binding of the neurotransmitter gamma-aminobutyric acid to its receptor, blocking the chloride ion uptake into the cells, and leading to hyperexcitation and death (Rhône-Poulenc, 1995).
The data related with genotoxicity of FP are very little in vertebrates, particularly in humans, in vitro and in vivo. Studies performed in rats explained that FP is quickly metabolized and residues are distributed in tissues, especially in fatty tissues under the skin and hair follicles (Hugnet et al., 1999). Ghisi et al. (2011) evaluated the genotoxicity of FP in Rhamdia quelen after subchronic contamination at three different concentrations (0.05, 0.1, 0.23 μg/L) using the comet and micronucleus (MN) test. They found that both 0.1 and 0.23 μg/L concentrations caused the most damage on DNA. These data suggest that FP may be highly carcinogenic to humans, despite being classified as class C—possible carcinogens (EPA, 2011a).
The comet assay, MN test, and sister chromatid exchange (SCE) analysis have been used to assess the toxicity and genotoxicity of many different chemicals, drugs, and pesticides in in vivo and in vitro studies in different organisms. The comet assay is capable of detecting DNA damage with great sensitivity and has been used widely both in in vitro and in vivo protocols to identify potentially environmental genotoxins (Tsuda et al., 2000). The comet assay has been widely accepted as a simple, sensitive, and rapid tool for assessing DNA damage and repair in individual eukaryotic as well as some prokaryotic cells, and has increasingly found application in diverse fields ranging from genetic toxicology to human epidemiology. The comet assay has been widely used in different models from bacteria to men, employing diverse cell types to assess the DNA-damaging potential of chemicals and/or environmental conditions by many research groups and researchers (Narendra, 2000; Sandal et al., 2008; Çavaş, 2011).
Genotoxic agents have the potential to interact with DNA and may cause DNA damage. SCE occurs spontaneously in proliferating cells and is regarded as a manifestation of damage to the genome. It has been commonly used as a test of mutagenicity to evaluate cytogenetic responses to chemical exposure, and dose–response relationships for different chemicals have been reported in both in vivo and in vitro studies (Bal et al., 1998; Çelik and Akbaş, 2005; Çelik and Aras Ateş, 2006). Countryman and Heddle proposed the human MN test in 1976 as a faster and alternative method to the metaphase analysis (Countryman and Heddle, 1976). The most popular type is the use of cytochalasin B to block cytokinesis, which causes the accumulation of binucleate cells at first mitosis (Fenech and Morley, 1985). There are, in fact, four recognized mechanisms by which MN and MN-like structures can arise: mitotic loss of acentric fragment, a variety of mechanical consequences of chromosomal breakage, an exchange or mitotic loss of whole chromosomes, and apoptosis. The latter is a form of nuclear destruction in which the nucleus disintegrates and nuclear fragments are formed (Tomanin et al., 1991; Koç-Başer et al., 1999). The simplicity of screening and scoring and the wide applicability of the in vitro MN test in different cell types make it a useful tool to evaluate cytogenetic damage (Kirsch-Volders et al., 2003). MN have been used as an indicator of chromosomal damage for more than 20 years (Chung et al., 2002).
This study was designed to evaluate the genotoxic and mutagenic potential of this compound in human peripheral blood lymphocytes exposed to different doses and demonstrate the damage caused by FP under in vitro conditions.
Materials and Methods
Chemical compound
FP, CAS number # 20068-37-3 and chemical name (RS)-5-aminno-1-(2,6-dichloro-α,α,α-trifluoro-p-toly)-4-trifluoro methylsulfinylpyrazole-3-carbonitrile was obtained from FIBREX 75 (5.5 mL contains 412.5 mg of FP).
Doses
FP doses used were based on an LD50 (for rats, 75 mg/kg). Final doses of FP administered in the experiments were obtained by dilution to 1 ‰ of LD50 (the highest dose)=0.7 μg/mL, 0.3 μg/mL (medium dose), and 0.1 μg/mL (the lowest dose). Mitomycin C and hydrogen peroxide (H2O2) were used as positive controls for SCEs, MN tests, and comet assay, respectively.
Subjects
This study was approved by the Clinical Researches Ethical Committee of Mersin University. Three healthy, male, nonsmoking donors (mean age, 29.32±2.33 years) provided blood samples. Subjects had not been exposed to radiation or drugs, 6 months before the study. Each person was interviewed about the possible influence of spurious factors.
Cytogenetic analysis
SCE analysis in lymphocyte cultures
All FP solution samples were sterilized using a Sartorius filter with a 0.22-μm-pore membrane. Three doses of FP were added to lymphocyte cultures under laboratory conditions. Lymphocyte cultures were prepared according to the technique of Moorhead et al. (1960) with slight modifications. Heparinized whole blood (0.8 mL) was added to 4.5 mL of culture medium F 10 (Gibco), supplemented with 20% of fetal calf serum (Gibco), 0.1% mL phytohemagglutinin (Gibco), and antibiotics (10,000 IU/mL penicillin and 10,000 IU/mL streptomycin). 5-Bromo-2-deoxyuridine (9 μg/mL; Sigma) was added to cultures at the beginning of the 72-h incubation period at 37°C for SCE analysis. Lymphocytes were cultured in the dark for 72 h, and metaphases were blocked during the last 1.5 h with colcemid at a final concentration of 0.2 μg/mL. Mitomycin C (2 μg/mL) was used as the positive control. The cells were harvested by replacing the culture medium with KCl (0.075 M) in which cells were incubated for 20 min at 37°C. The cells were fixed in the Carnoy's fixative (methanol:acetic acid, 3:1 v:v) five times, and slides were kept at room temperature overnight. Air-dried slides were stained according to the fluorescence-plus Giemsa method by Perry and Wolff (1974) with a slight modification. The number of SCEs was counted in 100 s—metaphase cells from each of the cultures on coded slides. Thus, 100 cells were scored in blind per culture for SCEs.
Cell proliferation kinetics and mitotic index
The mitotic index (MI) was calculated as the proportion of metaphases among the total cell population by counting a total of 1000 cells per culture. The cell proliferation kinetics (CPK) was defined as the proportion of the relative frequency of first division metaphases (M1, identifiable by uniform staining of both the sister chromatids), second division metaphases (M2, identifiable by differential staining of the sister chromatids), and third and subsequent division metaphases (M3, identifiable by nonuniform pattern of staining). The replication index (RI) or proliferation index (PRI) was calculated according to Ivett and Tice (1982). RI is the average number of replications completed by metaphase cells and is calculated as follows: RI=1×(% first division metaphases)+2×(% second division metaphases)+3×(% subsequent division metaphases)/100.
Cytokinesis block MN test
Blood samples were taken by venipuncture using heparinized vacutainers. Lymphocytes cultures were set up in the laboratory at a sterilized place and prepared according to the technique described by Scarpato et al. (1996) with slight modifications. Heparinized whole blood (0.8 mL) was added to 5 mL of culture medium, RPMI 1640 (Sigma), supplemented with 20% of fetal calf serum (Sigma), with 0.2 mL of phytohemagglutinin (Sigma), and with antibiotics (10,000 IU/mL penicillin and 10,000 IU/mL streptomycin). Three doses of FP (0.7, 0.3, and 0.1 μg/mL) were added in lymphocyte cultures. A final concentration of 6 μg/mL of cytochalasin B was added to cultures 44 h later to arrest cytokinesis. At 72 h of incubation, the cultures were harvested by centrifugation at 2000 rpm for 10 min. Then, to eliminate red cells and to keep the cytoplasm, the cell pellet was treated with a hypotonic solution (4–5 min 0.075 M KCl at 37°C). Cells were centrifuged, and the Carnoy's fixative (methanol:acetic acid, 3:1, v/v) solution was freshly added. This fixation step was repeated five times. Next, cell pellets were resuspended in a small volume of fixative solution and dropped onto clean, cold slides. The slides were stained with a 10% Giemsa dye solution.
Scoring criteria for MN and cytokinesis block proliferation index: To determine the frequency of binucleated cells with micronuclei (BNMN) and the total number of MN in lymphocytes, a total of 2000 binucleated cells with well-preserved cytoplasm were scored exactly for each subject and each dose on coded slides. MN were accepted only when (1) they were separated from the main nuclei, but included within the corresponding cytoplasm, (2) they had a chromatin material similar to that of the main nuclei, and (3) they were coplanar to the main nuclei. In the MN study, toxicity was evaluated by classifying cells according to the number of nuclei. The well-known cytotoxicity index was used: an index for measuring the CPK, called the cytokinesis block proliferation index (CBPI) (Fenech and Morley, 1985), which was calculated following the expression:
where MI–MIV represent the numbers of cells with one to four nuclei, respectively, and MIII and MIV are equally considered to be in their third cell cycle. As previously demonstrated, the CBPI is an accurate and biologically relevant index in detecting cellular toxicity or cell cycle delay (Fenech and Morley, 1985).
Blood sampling, cell preparation, and comet assay
The experiments were performed on peripheral blood lymphocytes obtained from three healthy donors, Peripheral blood mononuclear cells were isolated by Histopaque-1077 density gradient centrifugation, according to the manufacturer's instructions. Lymphocyte cultures were set up by adding 0.5 mL of lymphocyte suspension in 4.5 mL of RPMI 1640 medium supplemented with 20% of fetal calf serum, 2 mM
Alkaline comet assay: Comet assay was performed with lymphocytes of three donors according to Singh et al. (1988). Briefly, 100 μL of cell suspension was mixed with 200 μL of 2% low-melting-temperature agarose at 37°C and then placed on a slide precoated with a thin layer of 0.5% normal melting agarose. The cell suspension was immediately covered with a coverglass and the slides were kept at 4°C for 5 min to allow solidification of the agarose. After removing the coverglass, the cells were lysed in a lysing solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% Triton X-100, pH 10) for 1 h. After washing in redistilled water, the slides were placed in a horizontal gel electrophoresis chamber. The chamber was filled with a cold electrophoretic buffer (1 mM EDTA, 300 mM NaOH, pH 13) and slides were kept at 4°C for 40 min to allow the DNA to unwind. Electrophoresis was conducted at 20°C using 25 V and 185 mA for 20 min. After electrophoresis, the slides were washed three times with a neutralization buffer (0.4 M Tris, pH 7.5). All preparative steps were conducted in dark to prevent additional DNA damage. The slides were stained with with etidium bromide (0.1 mg/mL, 1:4) and analyzed with a fluorescence microscope (Olympus BX 51) equipped with a CCD-4230 A video camera.
Slide scoring: Fifty cells per slide and two slides were examined per sample to evaluate DNA damage for each FP concentration. The slides were blinded to the scorer. The cells were classified by eye into the five categories on the basis of the extent of DNA migration as undamaged (class 0), very little damage (class 1), moderate damage (class 2), high damage (class 3), and ultra high damage (class 4). The arbitrary unit (AU) was used to express the extent of DNA damage and calculated using the following formula.
N i =the number of scored cell in i level, i=the level of DNA damage (0, 1, 2, 3, 4).
Statistical analysis
Data were compared by analysis of variance (ANOVA). Statistical analysis was performed using the SPSS for Windows 9.05 package program. Post hoc analysis was performed by the least significant difference test. p<0.05 was considered as the level of significance.
Results
Table 1 represents the SCE and MN frequency and PRI, MI, and CBPI values of the control and each dose groups. All the doses of FP caused an increase in SCE and MN frequency and a decrease in PRI, MI, and CBPI values. There are significant differences between the control and FP dose groups except at the dose of 0.1 μg/mL for MN frequency (p<0.05). The mean of induced SCEs reached 5.80±0.08, 4.57±0.25, and 4.21±0.07 at the FP doses of 0.7, 0.3, and 0.1 μg/mL, respectively. Such values are much lower than those induced by the positive control, mitomycin C (8.50±0.24). Figures 1b and 1c show BNMN and SCE points in metaphase, respectively.
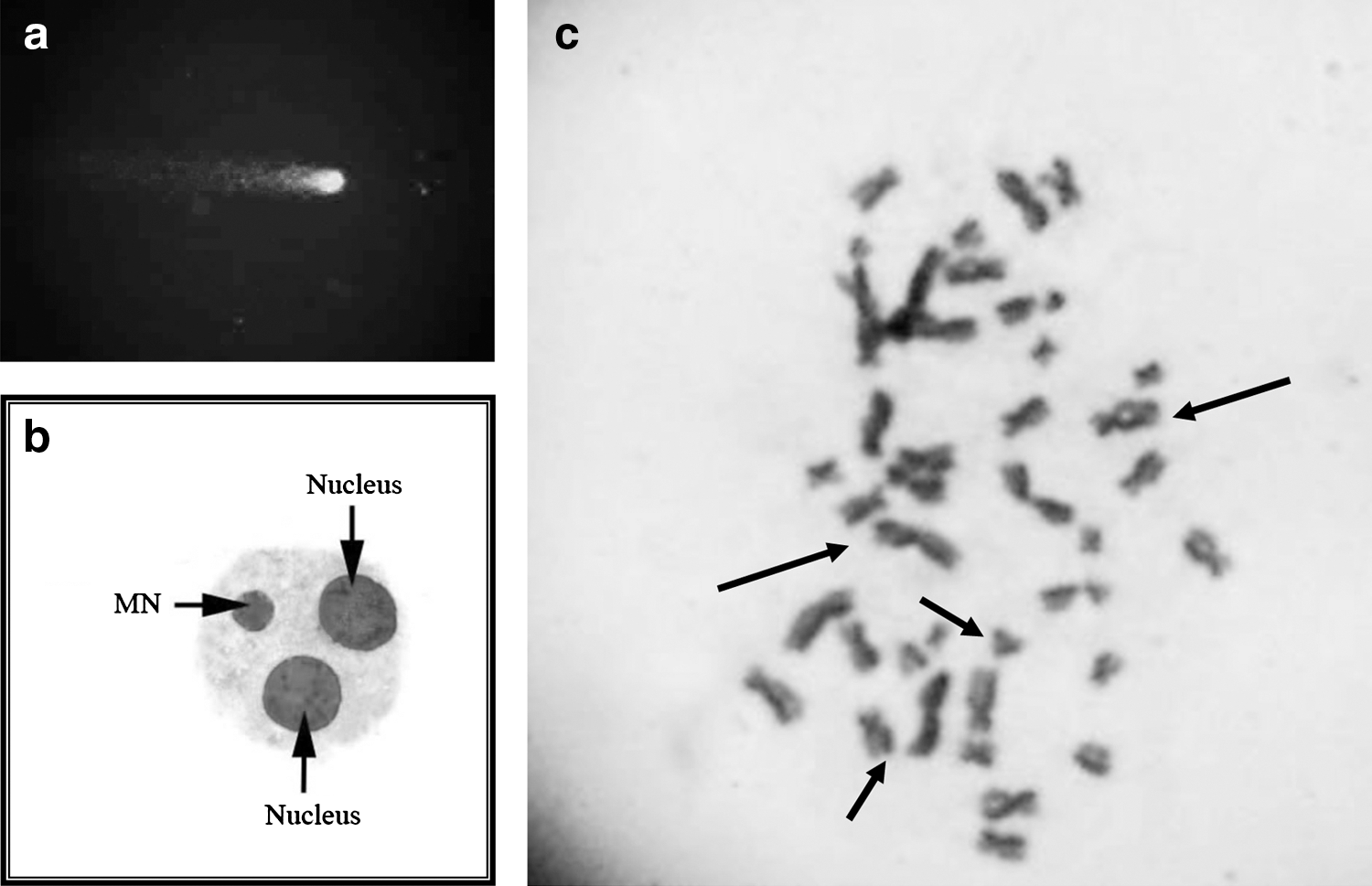
p<0.001.
p<0.01.
p<0.05 compared with negative control.
CBPI, cytogenesis block proliferation index; PRI, proliferation index; SCE, sister chromatid exchange; MN, micronucleus; PC, positive control; NC, negative control; SE, standart error; MI, mitotic index.
Table 2 shows the DNA damage level (class 0–class 4). All the doses of FP significantly induced DNA damage in dose-dependent manner in human peripheral blood lymphocytes (Fig. 1a). There is a significant difference between both dose groups and negative control and among dose groups (p<0.05).
p<0.05.
p<0.001 compared with negative control.
H2O2, hydrogen peroxide; TCS, total comet score.
Discussion
According to 2006–2007 market estimates, approximately 5.2 billion pounds of active pesticide ingredients are annually applied in the world (EPA, 2011b). Since 1945, more than 15,000 individual chemicals and 35,000 formulations have been used as agricultural pesticides (Forget et al., 1993). The chemicals are important tools for controlling agricultural pests, but they also represent a significant source of occupational and nonoccupational exposure to potentially toxic agents. Researches of other authors pointed out that some pesticides have genotoxic properties. Several studies demonstrated that pesticides can have genotoxic and cytotoxic effects in directly and indirectly exposed nontarget organisms (including humans) (Çelik et al., 2003; Çelik et al., 2005b, 2005c; Çavaş, 2011).
In vitro test systems or short-time tests are useful tools to examine the mechanism of action and standardize experimental stages. These tests are also capable of determining the potential hazard of compounds that are going to the marketplace, but are not required to be subjected to a full-scale toxicological assessment. Short-time tests identify the potential genotoxicity (Brusick, 1987).
The genotoxic effects of FP (active ingredient of FIBREX 75) were assessed using the comet assay and MN and SCE tests with human peripheral blood lymphocytes exposed to different doses of these chemicals in vitro.
SCE analysis, MN test, and comet analysis have been considered as efficient tools in the field of genetic toxicology in animals and humans (in vitro and in vivo, particularly, in occupational and biomonitoring) studies (Burgaz et al., 1998; Çelik et al., 2005a, 2013).
In the present study, mitomycin C is a compound widely used as a positive control in several studies in vitro and in vivo for SCE and MN tests (Çelik et al., 2003; Eke and Çelik, 2008) and H2O2 has been used as a positive control compound in several studies for comet assay and oxidative damage (Yuan et al., 2005; Sandal et al., 2008).
In our study, SCE analysis revealed that three doses of FP significantly induced SCE frequency (5.80±0.08, 4.57±0.25, and 4.21±0.07 and 0.7 μg/mL, 0.3 μg/mL, and 0.1 μg/mL, respectively) (p<0.001). Besides, MN analysis exhibited that the MN frequency increased in a dose-dependent manner. The dose 0.1 μg/mL of FP did not induced DNA damage in view of MN frequency (p>0.05). On the other hand, significant genotoxic effects were observed in the treatment with 0.7 and 0.3 μg/mL of FP (p<0.05, p<0.01 for 0.3, 0.7 μg/mL, respectively).
Since the development of the comet assay by Singh et al. (1988), it has been commonly utilized in research ranging from environmental biomonitoring to clinical diagnosis, to understand DNA repair processes and also in genetic toxicology (Rojas et al., 1999; Tice et al., 2000). Peripheral blood lymphocyte was used for the comet assay as it is the most preferred biomaterial for epidemiologic investigation, environment biomonitoring, in vitro genetic toxicology, and clinical study (evaluation of drug) (Rajaguru et al., 2002; Chen et al., 2008; Sandal et al., 2008).
Current published information on the genotoxic potential of methylpyrazole pesticides, particularly FP, is very limited. Ghisi et al. (2011) researched the effects of exposure of 60 days to the insecticide FP (concentrations of 0.05, 0.10, and 0.23 μg/L). They found that two concentrations—0.10 and 0.23 μg/L—of FP exposure caused the histopathological changes, nuclear morphological alterations, and DNA damage on the gills in R. quelen using the histopathologic analysis and the MN test/comet assay. They indicated that R. quelen may be a less sensitive bioindicator than other fish that have been tested.
In the present study, comet assay results showed that all the doses of FP induced DNA damage (DNA migration in comet assay) in the range of class 0–class 4. Graillot et al. (2012) observed an increased production of reactive oxygen species (ROS) in jurkat cells in the presence of four methylpyrazoles and demostrated that the four methylpyrazole pesticides (tebufenpyrad, bixafen, fenpyroximat, and tolfenpyrad) induced DNA damage in human cell lines. The relationship between the increase of DNA damage level in comet assay in this study and the increase in ROS level obtained in the study performed by Graillot is redolent of oxidative damage of DNA. Besides, Clasen et al. (2012) indicated that FP has potential toxic effects on nontarget organisms. They reported that FP insecticides cause alterations in the biochemical parameters in different tissues, such as the liver and brain, of the common carp without affecting growth during the 90-day exposure period. EFSA (2010) reported that tebufenpyrad showed some weak clastogenic potential in vitro. The experts agreed, however, when taking into account the entire database on genotoxicity and, in particular, the absence of a clastogenic potential in vivo, that overall, tebufenpyrad is devoid of genotoxic potency. It is indicated that fenproximate tested with various test systems (Ames test, DNA repair test, chromosomal aberration test, unsheduled DNA synthesis test) has no genotoxic potential in studies performed on living systems, including procaryotic and eucaryotic organisms (EFSA, 2010). Graillot et al. (2012) demonstrated that bixapen was genotoxic in jurkat cells with the same genotoxic potential as observed in SH-SY%Y cells. Huang et al. (2008) and Xia et al. (2008) indicated that different cells can show same effects against xenobiotics, including pesticides.
It is well documented that most major cellular responses observed both in vitro and in vivo after the exposure to chemicals, ionizing radiation, and/or other genotoxic agents are the inhibition or the delay of cell cycle progression. MI and RI or CBPI are used as indicators of sufficient cell proliferation and cytotoxicity (Anderson et al., 1988; Scott et al., 1991; Çelik, 2006; Eke and Çelik, 2008). The interaction between cytotoxicity and tumor promotion was indicated in several studies performed by many researchers (Albert and Magee, 2000). In addition, cytotoxicity causes the tumor promotion through inflammation and/or humoral immunity. FP exhibits a cytotoxic effect in vitro because of its inhibition of mitotic activity. The decrease in the mitotic activity is dose dependent. The frequency of MI decreased with increasing concentrations of FP (Table 1), and statistically significant differences from the control were observed, except at the dose of 0.1 μg/mL (p>0.05) compared with a negative control. Similar to our results, high cellular toxicity (decrease in MI) was recently reported in other cell culture models. Wang et al. (2013) used the insect cells (Sf9) derived from Spodoptera frugiperda pupal ovarian cells as an in vitro model. They reported that FPN was cytotoxic to Sf9 cells with a time- and concentration-dependent response. FP induced a Sf9 cell G2-M arrest and the percentage of G2-M phase cells evidently increased from 57.08%±0.29% to 90.48%±1.75%, while the proportion of apoptotic cells is also significantly increased from 4.48%±0.52% to 19.75%±1.01%. They observed the morphological changes, such as cell swelling, apoptotic bodies, and small fragments in Sf9 cells. These results indicated that FP could induce ROS production in Sf9 cells and then oxidative damage to DNA. The present study, based on an in vitro model using human peripheral blood lymphocyte cells, demonstrated that FP inhibited the cell division, decreasing the MI values. These results are on the same line with data of the study performed by Wang et al. (2013).
Further evaluation will be required to determine the relationship between toxic effects, genotoxic effects, and apoptotic effects, the DNA damage level, including oxidatively damaged bases, and the ROS level in human peripheral blood lymphocytes and different cell lines in vitro and in vivo. Clearly, there is a need for more detailed research on the effects of FP or its metabolites in the body, especially in the gene expression profile.
Footnotes
Disclosure Statement
No competing financial interests exist.