
Abstract
Viruses are intracellular parasites that must access the host cell machinery to propagate. Viruses hijack the host cell machinery to help with entry, replication, packaging, and release of progeny to infect new cells. To carry out these diverse functions, viruses often transform the cellular environment using viroporins, a growing class of viral-encoded membrane proteins that promote viral proliferation. Viroporins modify the integrity of host membranes, thereby stimulating the maturation of viral infection, and are critical for virus production and dissemination. Significant advances in molecular and cell biological approaches have helped to uncover some of the roles that viroporins serve in the various stages of the viral life cycle. In this study, the ability of viroporins to modify the cellular environment will be discussed, with particular emphasis on their role in the stepwise progression of the viral life cycle.
Introduction
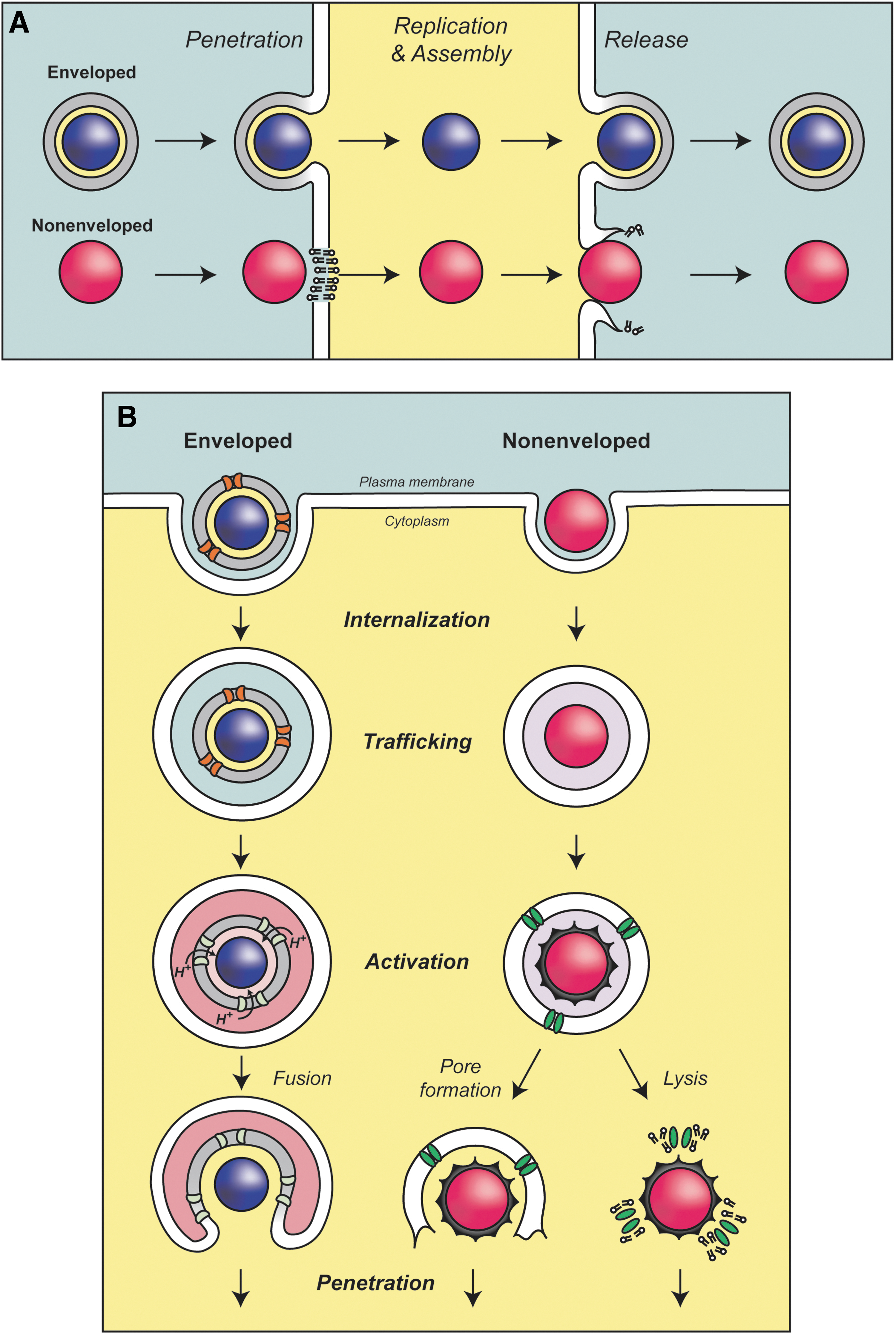
Models of viral infection and release.
N.D., not determined.
Entry and Penetration
Viruses bind to host cell receptors to initiate infection and entry (Marsh and Helenius, 2006). The mechanism used to overcome host cell membranes is dependent on the structural organization of the virus. For enveloped viruses such as the influenza virus and human immunodeficiency virus (HIV) that have a membrane bilayer, fusion with a host cell membrane delivers the viral particle into the cytoplasm during infection (Fig. 1A). In contrast, nonenveloped viruses contain a protein coat that surrounds their genome, so they must use a different strategy to translocate components across the bilayer during entry (Fig. 1A). Viroporins must be present in virions to assist with entry and penetration.
Influenza virus binds to the cell surface and is internalized into endosomes (Fig. 1B, left). The influenza virus viroporin M2 is located in the viral lipid bilayer and is critical for infection. M2 acts as a proton-conducting channel in the viral envelope that supports the acidification of the virus interior within endosomes (Pinto et al., 1992; Pielak and Chou, 2011; Wang et al., 2011). The envelope glycoprotein hemagglutinin undergoes a major conformational change that is triggered by the low pH of the endosome that leads to fusion of the viral envelope with the endosomal membrane and delivery of the nucleoprotein complex into the cytoplasm (Lakadamyali et al., 2003). The acidification by M2 is also thought to promote a structural rearrangement of the viral particle that is required for efficient uncoating of the viral RNA in the cytoplasm (Martin and Helenius, 1991). In the absence of M2, the viral protein capsid is delivered into the cytoplasm, but uncoating of the matrix protein M1 and the viral ribonucleoprotein does not occur and the infection is stalled (Watanabe et al., 2001; Takeda et al., 2002; Pinto and Lamb, 2007). The viroporin activity of M2 is inhibited by amantadine (Hay et al., 1985; Pinto et al., 1992; Wang et al., 1993). Chemically inhibiting the channel activity of M2 during infection prevents the infection, and amantadine was used as a treatment until drug-resistant mutations became prevalent (Davies et al., 1964; Bukrinskaya et al., 1982a, 1982b; Hay et al., 1985; Bright et al., 2006). Besides priming the virus particle for infection, M2 also serves other roles in the influenza virus life cycle that will be discussed later.
Several nonenveloped viruses also contain viroporins that are important for the penetration of host cell membranes. Viral proteins are frequently proteolytically cleaved during entry to release small hydrophobic viroporins that facilitate viral penetration. For example, μ1N is released from the nonenveloped reovirus capsid during infection (Fig. 1B, right) (Danthi et al., 2010). Viral particles containing a point mutation to inhibit the cleavage of μ1N entered the host cells but were noninfectious (Odegard et al., 2004). μ1N forms size selective pores in red blood cells and liposome membranes suggesting that it may also form pores during penetration that lead to disruption of the endosomal membrane for subviral particle release into the cytoplasm (Agosto et al., 2006). The adenovirus protein VI is also activated by proteolytic cleavage to aid penetration (Wiethoff et al., 2005). However, protein VI appears to have membrane disruptive properties that lyse the endosomal membrane by introducing positive membrane curvature rather than forming pores (Maier et al., 2010). In contrast, the VP4 viroporin from poliovirus is released by a structural rearrangement in the capsid after receptor binding (Danthi et al., 2003). Current results support the hypothesis that VP4 forms ion channels in the endosomal membrane that lead to lysis of the membrane for penetration into the cytoplasm, but further studies are needed to fully understand the viroporin-assisted penetration process.
The nonenveloped virus SV40 has a small double-stranded bidirectional genome that encodes for a total of seven proteins, of which four appear to possess the viroporin activity (Fiers et al., 1978; Daniels et al., 2006a, 2006b, 2007; Suzuki et al., 2010; Raghava et al., 2011; Giorda et al., 2012; Giorda et al., 2013; Raghava et al., 2013). To infect cells, SV40 is bound at the cell surface and endocytosed into caveolae-coated vesicles (Fig. 2, steps A–B) (Anderson et al., 1996; Pelkmans and Helenius, 2002; Pelkmans et al., 2005). The virus traffics to the to the endoplasmic reticulum (ER) using a microtubule-dependent mechanism (Fig. 2, steps C–D) (Pelkmans et al., 2001; Norkin et al., 2002). The ER contains chaperones and protein disulfide isomerases that induce structural rearrangements in VP1 that supports the uncoating and release of the minor structural proteins (Fig. 2, steps E–F) (Magnuson et al., 2005; Schelhaas et al., 2007; Geiger et al., 2011). The rearrangements that take place in the ER are likely required for penetration. The reorganized subviral particle is thought to be translocated across the ER membrane as a large intact viral particle (Inoue and Tsai, 2011). The delivery of the subviral particle into the cytoplasm would require subsequent targeting through nuclear pore complexes (Fig. 2, steps G–H). Alternatively, the ER is continuous with the nuclear envelope, so the delivery across the nuclear envelope could be more direct; however, there is no known pore in the inner nuclear membrane, although this route might be mediated by viral components (Fig. 2, step I) (Daniels et al., 2006b).
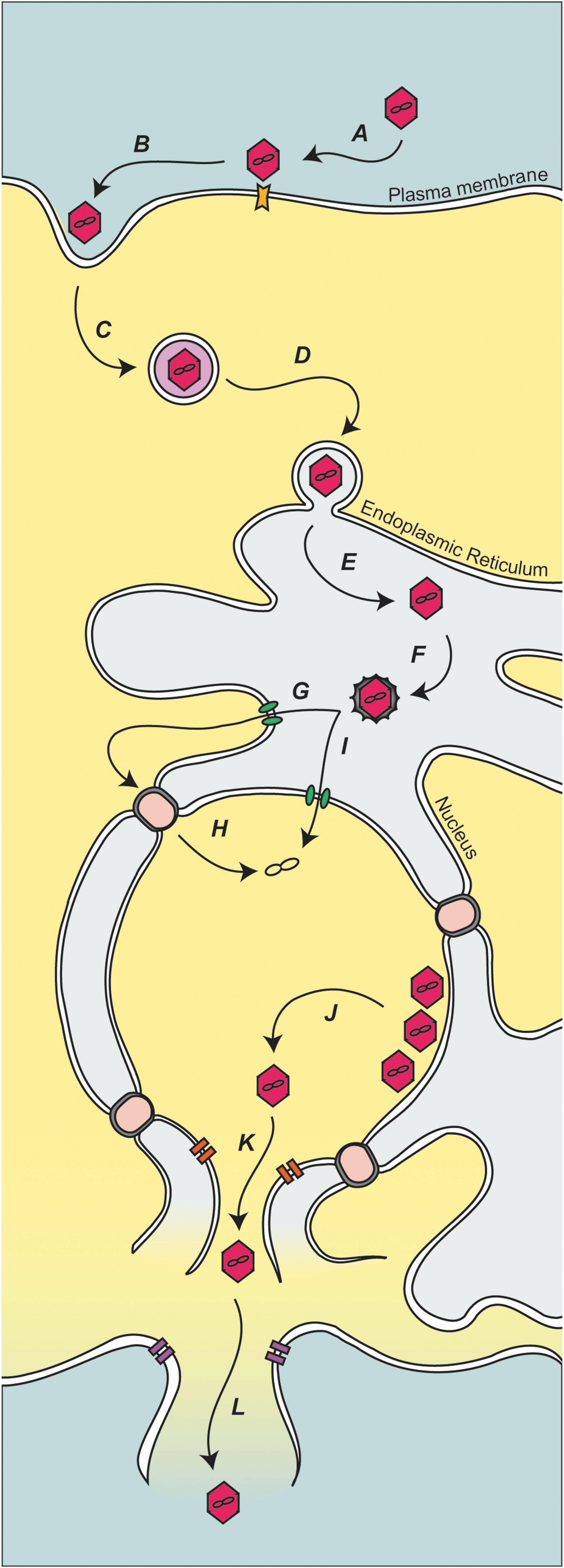
Model for polyomavirus infection and release. (A) Polyomavirus is bound at the plasma membrane by cellular receptors. (B) The virus is taken up by invagination of the plasma membrane. (C) The endocytosed virus is transported and (D) delivered to the ER. (E–F) Once the SV40 is in the ER, it is proposed that the viral protein coat is disassembled, releasing or exposing structural proteins VP2 and VP3. The released or exposed coat proteins aid viral transport to either the cytoplasm (G–H) or nucleus (I). (J) Capsid assembly around the viral minichromosome produces virions in the nucleus. (K) Newly produced VP4, along with (L) agnoprotein, triggers cytolysis of the host cell and release of the viral progeny. ER, endoplasmic reticulum.
VP2 and VP3 are required for efficient viral propagation (Daniels et al., 2006b). VP2 and VP3 could serve a role in penetration as in vitro-translated VP2 and VP3 insert into purified ER membranes. VP2 and VP3 form pores in model membranes, and mutant viruses lacking the pore formation activity are noninfectious (Giorda et al., 2013). The N-terminus of VP2 contains a conserved Glu residue that was critical for infection because it mediated an association to an ER-resident membrane protein, Bap31 (Geiger et al., 2011). Together, these results support the hypothesis that VP2 and VP3 act as soluble structural components during binding, entry, and assembly, and as membrane proteins for viral penetration. While some viruses utilize viroporins during penetration to modulate host membranes, the triggers used to activate the viroporins and the extent of membrane disruption are highly specialized.
Replication and Assembly
Following viral genome delivery to the host cell, some viruses modify the cellular environment to support viral genome replication. Two currently known viruses that require viroporins for replication include enteroviruses and coronaviruses. Viroporins from these viruses induce the dramatic remodeling of cellular membranes to create viral replication factories. These modified membranes originate from the secretory pathway and become sites of genome replication and assembly (Suhy et al., 2000; de Jong et al., 2006; Hsu et al., 2010b).
The best-characterized viroporin involved in genome replication is the enterovirus nonstructural protein 2B from poliovirus and coxsackievirus. The replication of the viral genome takes place on specialized vesicles that are derived from ER and Golgi membranes (van Kuppeveld et al., 1996; Sandoval and Carrasco, 1997). When expressed in cells, 2B appears to insert two transmembrane domains into ER membranes (Martinez-Gil et al., 2011). The oligomerization of 2B in the membrane forms pores that induce Ca+ leakage from the ER lumen and inhibits vesicular transport (Barco and Carrasco, 1995; Doedens and Kirkegaard, 1995; Aldabe et al., 1997; de Jong et al., 2008). 2B also contributes to the virus release by increasing the permeability of the plasma membrane. The current hypothesis is that the level of 2B production in the cell dictates the localization and function of the protein to aid either in replication or release (Martinez-Gil et al., 2011).
The viroporin p7 is essential for hepatitis C virus infectivity (Sakai et al., 2003; Jones et al., 2007). The channel activity of bacterially expressed and purified p7 is inhibited with amantadine (Griffin et al., 2003). Hepatitis C virus genome replication takes place in the cytoplasm and coincides with membrane remodeling, including the formation of lipid droplets that originate from the ER. Lipid droplets help to enhance the efficiency of virus assembly by concentrating viral core proteins in close proximity to structural components and viral genomes that are situated in ER membranes (Boson et al., 2011). The p7 viroporin functions as a calcium channel and interacts with the nonstructural protein 2 (NS2) (Griffin et al., 2003; Tedbury et al., 2011). p7 is not required for viral genome replication, but is essential for subsequent steps in virus assembly (Jones et al., 2007). The basic amino acid residues that are critical for channel function are important for the release of infectious virions (Griffin et al., 2004; Jones et al., 2007). Together, p7 and NS2 appear to coordinate the hand off of the viral core from lipid droplets to ER sites for viral assembly (Boson et al., 2011).
Some envelope glycoproteins induce fusion of the viral envelope with the host cell membrane to support viral penetration (Fig. 1B, left). Many viruses are internalized to endosomal compartments where they are activated by the exposure to acidic pH. Influenza hemagglutinin undergoes a major conformational change upon exposure to acidic conditions that mediates membrane fusion (Harrison, 2008). The glycoproteins must be kept in a neutral pH environment when maturing in the secretory pathway to prevent premature activation. Both M2 and p7 appear to disrupt the proton gradient in the secretory pathway to aid the maturation of active virus particles (Sugrue et al., 1990; Sugrue and Hay, 1991; Jones et al., 2007). A channel inactive mutant of hepatitis C virus p7 can be rescued by the expression of influenza virus M2 or using bafilomycin A1 to chemically inhibit endosome acidification (Wozniak et al., 2010). M2 and p7 appear to disrupt the pH gradient in the secretory pathway so that active viruses are released from infected cells (Sugrue et al., 1990; Sugrue and Hay, 1991; Jones et al., 2007). Altogether these results demonstrate that viroporins contribute to the viral replication and assembly processes by modifying the cellular environment and coordinating interactions with other viral proteins.
Release
Membrane budding and scission events are commonly used to release enveloped viruses from infected cells (Fig. 1A, top right). Several enveloped viruses exploit the ESCRT pathway of host cells to complete membrane fission at the virus bud neck (Chen and Lamb, 2008). Influenza virus appears to use M2 for membrane fission instead of the ESCRT pathway as M2 induces the membrane invagination of liposomes and the pinching off of membrane vesicles (Rossman et al., 2010). A conserved amphipathic domain is required for this activity as its deletion inhibits the release of viral particles from host cells indicating that M2 plays a direct role in membrane fission for virus spread.
The alphavirus 6K viroporin conducts ions across planar lipid bilayers (Melton et al., 2002). The synthesis of 6K in Escherichia coli and mammalian cells induces toxicity and permeability, respectively (Sanz et al., 1994; Joe et al., 1998). 6K is produced as part of a polyprotein that is generated after cleavage and traffics as a complex to the plasma membrane for the assembly and release of the enveloped virus (Liljeström and Garoff, 1991). The 6K protein is associated with the envelope glycoproteins throughout the secretory pathway and may enhance their trafficking (Gaedigk-Nitschko et al., 1990; Lusa et al., 1991; Loewy et al., 1995). In the absence of 6K, the viral genome is replicated, and the synthesis and intracellular trafficking of envelope proteins is detected; however, virus particles do not efficiently bud from cells (Bredenbeek et al., 1993; Loewy et al., 1995). 6K appears to directly modify the lipid bilayer to prompt virus release potentially by depolarizing the plasma membrane (Sanz et al., 2003).
Enveloped coronaviruses use the E protein viroporin in viral assembly and release from host cells (Corse and Machamer, 2000; Lim and Liu, 2001; Madan et al., 2005). E protein expressed in E. coli or mammalian cells increases the permeability of the plasma membrane (Liao et al., 2004; Madan et al., 2005). It is found at low levels in the viral envelope and is important for the release of infectious bronchitis virus (Machamer and Youn, 2006).
Vpu is found in the more virulent human immunodeficiency virus type 1 (HIV-1) and is absent from type 2 (HIV-2). It is a cation selective ion channel when expressed in xenopus oocytes (Schubert et al., 1996). The expression of Vpu in mammalian cells or E. coli causes increased permeability to small molecules (Gonzalez and Carrasco, 1998). This led to the hypothesis that the function of Vpu during viral release is to depolarize the plasma membrane to enhance the release of viral particles (Hsu et al., 2010a). HIV-2 encodes for two alternative ion conducting channel proteins (ROD10 and ST2) that appear to substitute for the function of Vpu in release (Bour et al., 1996; Ritter et al., 1996; Bour and Strebel, 2003). The depolarization of the plasma membrane is thought to enhance the fusion of the membrane around the capsid and assist in the progression of the membrane fission reaction (Hsu et al., 2010a).
Nonenveloped viruses are commonly released through a lytic mechanism so that viruses are free of membranes (Fig. 1A, bottom right). The bluetongue virus nonstructural protein 3 (NS3), poliovirus 2B, and JC virus agnoprotein increase the membrane permeability of mammalian cells and are associated with viral release (Doedens and Kirkegaard, 1995; Aldabe et al., 1996; Han and Harty, 2004; Suzuki et al., 2010). 2B and agnoprotein traffic through the ER and Golgi and eventually permeabilize the plasma membrane by forming small pores (Agirre et al., 2002; Suzuki et al., 2010). NS3, 2B, and agnoprotein appear to directly assist in nonenveloped virus release by forming pores in the plasma membrane that lead to timely cell lysis.
SV40 viral progeny are assembled in the nucleus of infected cells (Fig. 2, step J). A number of results support a viroporin role in stimulating viral release for a recently discovered protein produced by SV40 termed VP4: (1) VP4 expression in E. coli increases membrane permeability; (2) VP4 is expressed when infected host cells become permeable; (3) deletion of VP4 from the viral genome slows the rate of viral propagation; (4) purified bacterial expressed VP4 forms size selective pores in model membranes; (5) when expressed in host cells, VP4 is localized along the nuclear envelope and disrupts membrane integrity; (6) like other viroporins, VP4 requires a basic amino acid sequence for the membrane disruption activity; and (7) fluorescent-based assays indicate that VP4 induces transbilayer diffusion of lipids through a toroidal pore structure (Daniels et al., 2007; Raghava et al., 2011; Giorda et al., 2012; Raghava et al., 2013). A viroporin activity has also been described for agnoprotein from JC polyomavirus that disrupts the plasma membrane of infected cells to promote virus release (Suzuki et al., 2010). As the polyomavirus SV40 also encodes for agnoprotein, this presents the possibility that VP4 and agnoprotein work in concert during virus release to disrupt both the nuclear envelope and plasma membrane (Fig. 2 steps K–L).
Perspectives
Viruses are able to subvert cellular processes and modify the cellular environment to reproduce. Viroporins are ingeniously complicated, small, and efficient proteins that dramatically affect the host cell. Future work should aim to identify new viroporins and understand how they modify the cellular environment. The approaches used to study p7, M2, Vpu, and SV40 VP2-4 provide a framework that can be applied to the study of other viroporins. A deeper understanding of the full range of viroporins and their roles in the viral life cycle could be leveraged to create pharmacological agents that inhibit viral replication or engineer pseudoviruses as gene delivery vehicles. Viroporin inhibitors might also be used to uncover the stage of the viral life cycle that channel activity is required. The application of molecular biology, biochemical, structural, and cell biology methodologies to the study of viroporins will greatly enhance our understanding of these resourceful proteins. Detailed analysis of viroporins throughout the virus maturation process provides valuable insight into the virus and fundamental cell biology.
Footnotes
Acknowledgments
Related work was supported by a grant from the National Institutes of Health (AI078142 to D.N.H.). K.M.G. was partially supported by the National Science Foundation, Integrative Graduate Education and Research Traineeship (IGERT), the Institute for Cellular Engineering (DGE-0654128), and the National Institutes of Health Chemistry-Biology Interface training grant (T32GM00815).
Disclosure Statement
No competing financial interests exist.