
Abstract
Congenital heart disease (CHD) is the most common form of birth defect and is the leading noninfectious cause of infant death. A growing body of evidence demonstrates that genetic risk factors are involved in the pathogenesis of CHD. However, CHD is a genetically heterogeneous disease and the genetic defects underlying CHD in an overwhelming majority of patients remain unclear. In this study, the whole coding region and splice junction sites of the PITX2c gene, which encodes variant 3 of paired-like homeodomain transcription factor 2 crucial for normal cardiovascular morphogenesis, were sequenced in 382 unrelated patients with CHD, and 2 novel heterozygous mutations, p.W147X and p.N153D, were identified in 2 unrelated patients with CHD, respectively, including a 1-year-old male patient with double outlet right ventricle in combination with ventricular septal defect and a 4-year-old female patient with ventricular septal defect. The mutations were absent in 400 control chromosomes and were both predicted to be disease-causing by MutationTaster. Multiple alignments of PITX2c proteins across species displayed that the altered amino acids were completely conserved evolutionarily. Functional analysis revealed that the mutated PITX2c proteins were associated with a significantly reduced transactivational activity compared with their wild-type counterpart. These findings provide a novel insight into the molecular mechanisms implicated in CHD, suggesting potential implications for the antenatal prophylaxis and allele-specific treatment of CHD.
Introduction
Embryonic heart development is an exquisitely complex and dynamic biological process that requires the orchestration of cardiac cell commitment, differentiation, proliferation, and migration, which is meticulously regulated by an evolutionarily conserved network of transcription factors that connect signaling pathways with genes for muscle growth, patterning, and contractility (Pikkarainen et al., 2004; Olson, 2006; Bruneau, 2008; Bartlett et al., 2010; Cecchetto et al., 2010), and both genetic and environmental risk factors may perturb the exquisite and coordinated interactions of transcription networks that govern cardiovascular morphogenesis, possibly leading to embryonic lethality or CHD (McCulley and Black, 2012). Moreover, mutations in multiple cardiac transcription factor genes, including GATA4, GATA5, GATA6, and NKX2-5, have been implicated in the pathogenesis of CHD (Schott et al., 1998; Garg et al., 2003; Reamon-Buettner and Borlak, 2010; Wang et al., 2012; Zheng et al., 2012; Fahed et al., 2013; Huang et al., 2013; Jiang et al., 2013; Wei et al., 2013a, 2013b; Yuan et al., 2013). However, CHD is a genetically heterogeneous disease and the molecular defects responsible for CHD in an overwhelming majority of patients remain to be identified.
Recently, a growing body of evidence indicates that the cardiac transcription factor PITX2c, a member of the paired-like homeobox family of transcription factors, plays a key role in cardiovascular genesis (Liu et al., 2002; Dagle et al., 2003; Bamforth et al., 2004; Mommersteeg et al., 2007; Galli et al., 2008; Lozano-Velasco et al., 2011). In human, the PITX2c gene is predominantly expressed in the embryonic and adult hearts (Clauss and Kääb, 2011), and is crucial for the normal embryonic development of left atrium, cardiac conduction system, and pulmonary venous myocardium (Douglas et al., 2011). In mice, deletion of PITX2c has been associated with a wide variety of CHDs, including atrial isomerism, double-outlet right ventricle, atrial septal defect, ventricular septal defect, transposition of the great artery, persistent truncus arteriosus, and abnormal aortic arch as well as incomplete closure of the body wall (Gage et al., 1999; Lin et al., 1999; Liu et al., 2001). These findings justify screening PITX2c as a prime candidate gene for CHD.
Materials and Methods
Study subjects
A cohort of 382 unrelated patients with CHD was recruited from the Chinese Han population. The available relatives of the index patients carrying the identified PITX2c mutations were also enlisted. Subjects were evaluated by individual and familial histories, review of the medical records, complete physical examination, standard 12-lead electrocardiogram, and two-dimensional transthoracic echocardiography with color flow Doppler. The types of CHD were determined using two-dimensional continuous wave Doppler and color Doppler techniques on transthoracic echocardiography. Where necessary, transesophageal echocardiography and angiography were used for further clarification of the anatomy. Some patients underwent cardiac catheterization and, if required, cardiac surgery. The patients with known chromosomal abnormalities or syndromic cardiovascular defects were excluded from the study.
A total of 200 ethnically matched, unrelated healthy individuals randomly selected from those undergoing routine physical examinations were used as controls. Based on reviews of medical histories and analyses of echocardiographic records, the control individuals had no congenital cardiovascular anomalies, except for subclinical cardiac aberrations such as a bicuspid aortic valve or a patent foramen ovale. The ethnic origin of the participants was determined by a combination of self-reported ethnicity and a personal questionnaire asking questions about the birthplace, language, religion, and ancestry.
Peripheral venous blood samples from CHD patients and control individuals were prepared. The study protocol was reviewed and approved by the local institutional ethics committee and written informed consent was obtained from all participants or their guardians before the study.
Genetic studies
Genomic DNA from all participants was extracted from blood lymphocytes with the Wizard Genomic DNA Purification Kit (Promega, Madison, WI). The whole coding region and splice junction sites of the PITX2c gene were initially sequenced in 382 unrelated patients with CHD and subsequently genotyped for the available relatives of the probands carrying identified mutations and the 200 control individuals. The referential genomic DNA sequence (accession No. NC_000004) and RNA sequence (accession No. NM_000325) of PITX2c were derived from GenBank, which was at the National Center for Biotechnical Information (NCBI;
Alignment of multiple PITX2c protein sequences among species
Alignment of multiple PITX2c protein sequences across various species were carried out using the online program of MUSCLE, version 3.6 (
Prediction of the causative potential of a PITX2c sequence variation
The disease-causing potential of a PITX2c sequence variation was predicted by MutationTaster (an online program at
Plasmids and site-targeted mutagenesis
The recombinant expression plasmid PITX2c-pcDNA4, which was constructed by Strungaru et al. (2011), was a gift from Prof. Georges Christé, from Physiopathologie des Troubles du Rythme Cardiaque, Faculté de Pharmacie de Lyon, Université Lyon 1, Lyon, France. The atrial natriuretic factor (ANF)-luciferase reporter plasmid, which contains the 2600-bp 5′-flanking region of the ANF gene, namely ANF(-2600)-Luc, was kindly provided by Dr. Ichiro Shiojima, from the Department of Cardiovascular Science and Medicine, Chiba University Graduate School of Medicine, Chuo-ku, Chiba, Japan. The identified mutation was introduced into the wild-type PITX2c using a QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) with a complementary pair of primers. The mutant was sequenced to confirm the desired mutation and to exclude any other sequence variations.
Transcriptional activity assay
Chinese hamster ovary (CHO) cells were cultured in the Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum as well as 100 units/mL penicillin and 100 μg/mL streptomycin. The ANF(-2600)-Luc reporter construct and an internal control reporter plasmid pGL4.75 (hRluc/CMV; Promega) were used in transient transfection experiments to assay the transactivational activity of the PITX2c mutants. CHO cells were transfected with 2 μg of wild-type PITX2c–pcDNA4 or mutant PITX2c–pcDNA4 (W147×or N153D) or empty vector pcDNA4, 2.0 μg of ANF(-2600)-Luc reporter construct, and 0.04 μg of pGL4.75 control reporter vector. For cotransfection experiments, 1 μg of wild-type PITX2c–pcDNA4, 1 μg of mutant PITX2c–pcDNA4 (W147×or N153D), 2.0 μg of ANF(-2600)-Luc, and 0.04 μg of pGL4.75 were used. Transient transfection was performed 24 h after plating with Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA). The transfected cells were incubated for 24 h, then lysed, and assayed for reporter activities. Firefly luciferase and Renilla luciferase activities were measured with the Dual-Glo luciferase assay system (Promega). The activity of the ANF promoter was presented as fold activation of Firefly luciferase relative to Renilla luciferase. Three independent experiments were performed at minimum for wild-type and mutant PITX2c.
Statistical analysis
Data are expressed as means±standard deviations. Continuous variables were tested for normality of distribution, and the Student's unpaired t-test was used for comparison of numeric variables between two groups. The Pearson's χ 2 test or Fisher's exact test was used, when appropriate, to compare the categorical variables between two groups. A two-tailed p<0.05 indicated statistical significance.
Results
Clinical characteristics of the study population
A cohort of 382 unrelated patients with CHD was clinically evaluated in contrast to a total of 200 ethnically matched, unrelated healthy individuals. None of them had apparent environmental risk factors for CHD, such as maternal illness and drug use in the first trimester of pregnancy, parental smoking, and chronic exposure to toxicants and ionizing radiation. The baseline clinical characteristics of the 382 patients are summarized in Table 1.
CHD, congenital heart disease; ASD, atrial septal defect; VSD, ventricular septal defect; TOF, tetralogy of Fallot; PDA, patent ductus arteriosus; DORV, double outlet right ventricle; PS, pulmonary stenosis; TAPVC, total abnormal pulmonary venous connection; COA, coarctation of the aorta; TGA, transposition of the great arteries; CAVC, common arteriovenous canal; PTA, persistent truncus arteriosus; PFO, patent foramen ovale; AF, atrial fibrillation.
Identification of PITX2c sequence variations
Two heterozygous sequence variations in PITX2c were identified in 2 of 382 patients with CHD, respectively, with a mutational prevalence of ∼0.52%. Specifically, a substitution of adenine for guanine at coding nucleotide 441 of the PITX2c gene (c.441G>A), equivalent to the replacement of tryptophane by stop codon at amino acid position 147 (p.W147X), predicting the truncated protein without C-terminal 178 amino acids, was identified in the proband from family 1, a 1-year-old male patient with double outlet right ventricle in combination with ventricular septal defect. A transition of adenine to guanine in the first nucleotide of codon 153 of the PITX2c gene (c.457A>G), resulting in the transversion of asparagine to aspartic acid at amino acid 153 (p.N153D), was identified in the proband from family 2, a 4-year-old female patient with ventricular septal defect. The sequence chromatograms showing the detected heterozygous PITX2c variations in contrast to corresponding control sequences are shown in Figure 1. A schematic diagram of PITX2c showing the structural domains and the locations of the identified mutations is presented in Figure 2. The variants were neither observed in 400 control alleles nor found in the EVS and SNP databases, which were consulted again on August 3, 2013.
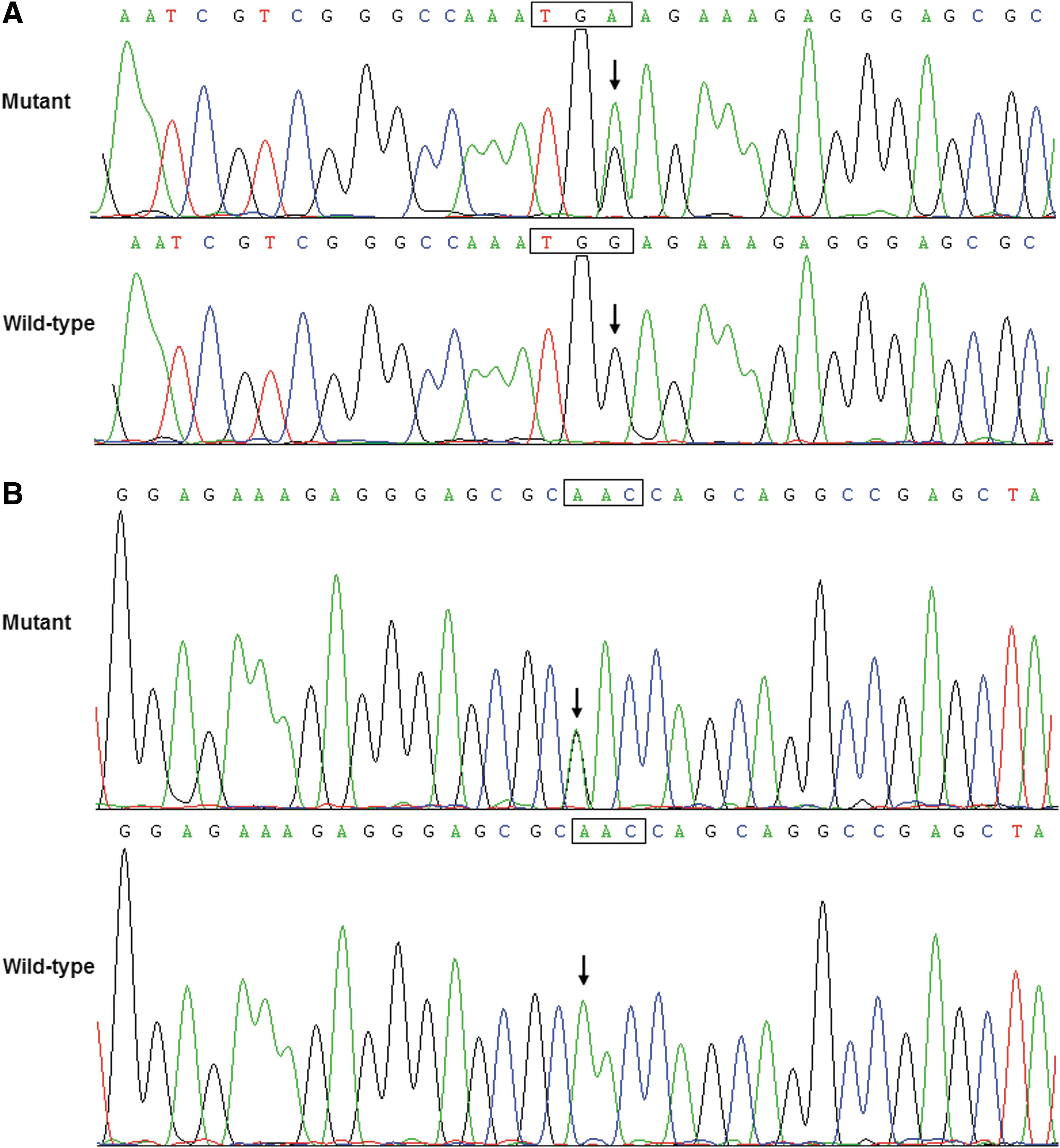
Sequence electropherograms showing the PITX2c mutations in contrast to their corresponding controls. The arrow indicates the heterozygous nucleotides of G/A in the probands from families 1

Schematic diagram of the PITX2c protein structure with the CHD-related mutations indicated. The mutations identified in patients with CHD are shown above the structural domains. NH2, aminoterminus; TAD1, transcriptional activation domain 1 (amino acids 1–91); HD, homeodomain (amino acids 92–151); NLS, nuclear localization signal (amino acids 145–161); TID1, transcriptional inhibitory domain 1 (amino acids 162–212); TAD2, transcriptional activation domain 2 (amino acids 213–285); TID2, transcriptional inhibitory domain 2 (amino acids 286–324); COOH, carboxylterminus; CHD, congenital heart disease.
A genetic screen of the mutation carriers' family members demonstrated that in family 2, the variation was present in all affected family members available, but absent in unaffected family members examined; while in family 1, the variation was only present in the proband (III-2), and absent in his unaffected parents, indicating a de novo mutation. Analysis of the pedigrees showed that in family 2, the variation cosegregated with CHD with complete penetrance. The pedigree structures of the two families are illustrated in Figure 3. All the affected family members from both families had the same anatomic type of subarterial ventricular septal defect. The index patient from family 1 (III-2) had also double outlet right ventricle and the proband's grandfather from family 2 (I-1) had also documented atrial septal defect. The phenotypic characteristics and results of genetic screening of the affected pedigree members are listed in Table 2.
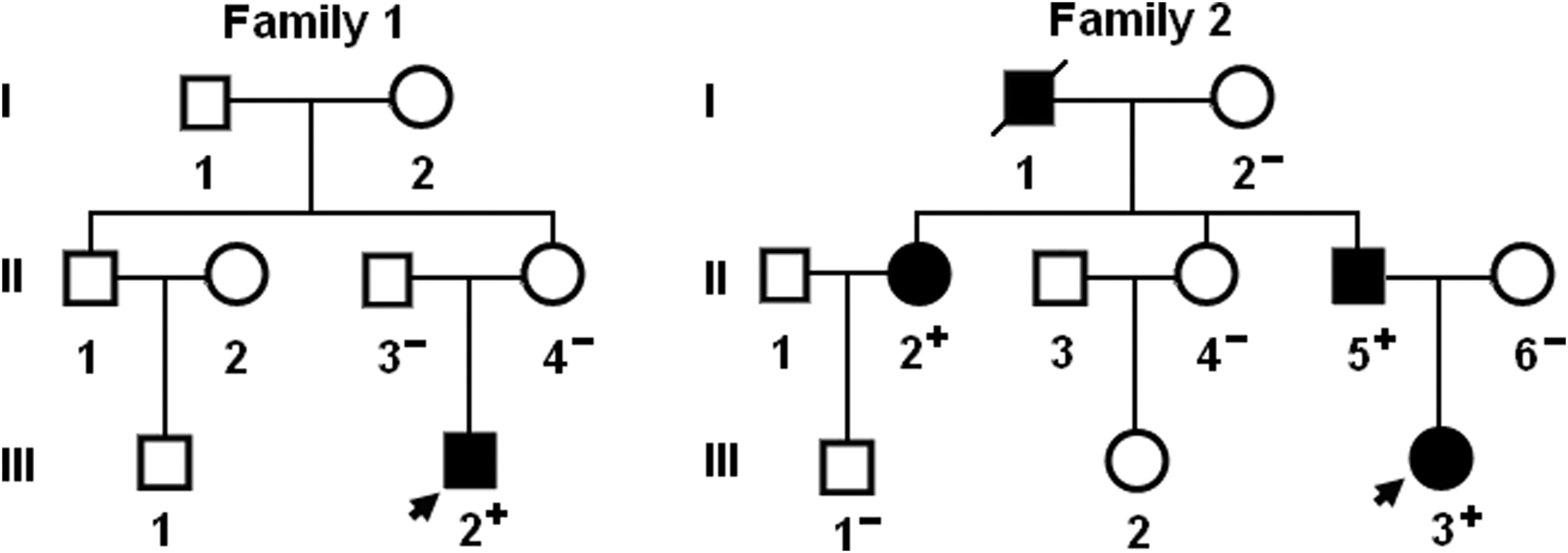
Pedigree structures of the families with CHD. Families are designated as family 1 and family 2, respectively. Family members are identified by generations and numbers. Squares indicate male family members; circles, female members; closed symbols, affected members; open symbols, unaffected members; arrows, probands; a symbol with a slash, the deceased member; “+,” carriers of the heterozygous mutations; and “−,” noncarriers.
Age at death.
+, present; −, absent; M, male; F, female; NA, not available or not applicable.
Multiple alignments of PITX2c sequences across species
As shown in Figure 4, a cross-species alignment of multiple PITX2c protein sequences displayed that the affected amino acids were completely conserved evolutionarily, indicating that the amino acids are functionally important.

Alignment of multiple PITX2c protein sequences across species. The altered amino acids of p.W147 and p.N153 are completely conserved evolutionarily.
Disease-causing potential of PITX2c sequence variations
The PITX2c sequence variations of c.441G>A and c.457A>G were both automatically predicted to be disease-causing, with the same p-value of 1.00000. No SNPs in the altered regions were found in MutationTaster database.
Transcriptional activity of the PITX2c mutants
The wild-type PITX2c, the W147X-mutant, and the N153D-mutant PITX2c activated the ANF promoter by ∼10-, ∼3-, and ∼4-fold, respectively. When wild-type PITX2c was coexpressed with the same amount of W147X-mutant or N153D-mutant PITX2c, the induced activation of the ANF promoter was ∼7- or ∼5-fold. These results reveal that both PITX2c mutants are associated with significantly reduced activation activity compared with their wild-type counterpart (Fig. 5).

Functional impairments resulted from PITX2c mutations. Activation of ANF promoter driven luciferase reporter in CHO cells by PITX2c WT, W147X-mutant, or N153D-mutant, alone or in combination, demonstrated a significantly reduced transactivational activity by mutant proteins. Experiments were performed in triplicate, and mean and standard deviations are shown. **p<0.001 and *p<0.05, when compared with the same amount (2 μg) of wild-type PITX2c. ANF, atrial natriuretic factor; CHO, Chinese hamster ovary; WT, wild-type.
Discussion
In the present study, two novel heterozygous mutations of PITX2c, p.W147X and p.N153D, were identified in two families with CHD, respectively. The mutant alleles were present in the affected family members available, but absent in unaffected relatives examined and 400 reference chromosomes from an ethnically matched control population. A cross-species alignment of PITX2c protein sequences displayed that the altered amino acids were completely conserved evolutionarily. These two mutations were both predicted to be causative, and the functional analysis revealed that the mutant PITX2c proteins were associated with a significantly reduced transactivational activity. Therefore, it is likely that functionally impaired PITX2c predisposes to CHD in these families.
PITX2 belongs to the bicoid-like homeodomain transcription factor family. To date, four different PITX2 transcripts, generated by differential mRNA splicing and alternative promoter usage, have been identified, of which PITX2a, PITX2b, and PITX2c differ only in their amino-termini and are expressed in human, mice, chick, zebrafish, and Xenopus, whereas the fourth isoform, PITX2d, which lacks the amino-terminal domain and most of the homeodomain, is only detected in humans. The unique amino-termini of PITX2a, PITX2b, and PITX2c modify their ability to activate transcription in a cell-type and promoter-dependent manner. The homeodomain is responsible for recognizing a specific DNA sequence 5′-TAATCC-3′, which is required for DNA binding and interaction with other transcription factors. The nuclear localization signal is associated with subcellular trafficking (Simard et al., 2009). In human, PITX2c gene maps to chromosome 4q25, encoding a protein of 324 amino acids (Semina et al., 1996), and is expressed asymmetrically in the developing and adult heart, playing an important role in cardiovascular morphogenesis and maturation (Kirchhof et al., 2011). The PITX2c mutations p.W147X and p.N153D identified in this study are located in homeodomain and nuclear localization signal, respectively, and the p.W147X mutation eliminates the partial homeodomain, nuclear localization signal, and the entire C-terminus, and thus may be expected to perturb the transcriptional activation of PITX2c by interfering with the binding of PITX2c to target gene promoter or nuclear translocation.
Previous studies have established PITX2c as an upstream regulator of several target genes expressed in the heart during embryogenesis, including the gene coding for ANF (Ganga et al., 2003). Thus, the functional role of a PITX2c mutation can be explored by an assay of the transcriptional activity of the ANF promoter in tool cells. In this study, the functional characteristics of two novel PITX2c mutations identified in CHD patients were delineated by transcriptional activity analysis and the results unveiled that the two mutations were both associated with a significantly reduced transcriptional activity on a downstream gene ANF, implying that functionally compromised PITX2c is potentially an alternative pathological mechanism of CHD.
Association of functionally impaired PITX2c with increased susceptibility to congenital cardiovascular deformations has been substantiated in animal models. In mice, PITX2c was expressed predominantly in the trabecular and septal myocardium with a strong expression bias in the myocardium associated with endocardial cushions of the atrioventricular canal and outflow tract (Furtado et al., 2001), and knockout of the PITX2c gene resulted in embryonic death, due to cardiovascular defects, including double-outlet right ventricle, atrial isomerism, atrial septal defect, ventricular septal defect, and abnormal aortic arch (Liu et al., 2001). In Xenopus, knockdown of PITX2c by RNA interference led to defects in the cardiac outflow tract and cardiac septation (Dagle et al., 2003). These experimental findings highlight an exquisite sensitivity of the developing cardiovascular system to the level of PITX2c.
It has been demonstrated that during embryogenesis, multiple genes are transactivated by PITX2c (Clauss and Kääb, 2011), and genetic mutations in several downstream target molecules have been linked to CHD, including GATA4, NKX2.5, CX40, and CX43 (Britz-Cunningham et al., 1995; Schott et al., 1998; Garg et al., 2003; Krüger et al., 2006; Soemedi et al., 2012). Hence, mutated PITX2c may enhance the vulnerability to CHD by decreasing the expressions of such cardiac-specific target genes.
In addition, by exome sequencing, Zaidi et al. (2013) previously screened 362 patients with sporadic CHD, including 154 with conotruncal defects, 132 with left ventricular obstruction, 70 with heterotaxy, and 6 with other cardiac structural deformations, and found a de novo mutation in PITX2c, p.A47V, in a patient with left ventricular obstruction (coarctation of the aorta), with a mutational prevalence of roughly 0.28%. However, the functional characteristics of the PITX2 mutation associated with idiopathic coarctation of the aorta remain to be delineated.
It was interesting that PITX2c mutations were related to lone atrial fibrillation (AF). Yang et al. (2013) genotyped PITX2c in 152 unrelated index patients with familial AF and their available relatives, and identified two novel heterozygous PITX2c mutations, p.S37W and p.Y280X, in two families with AF, respectively. Zhou et al. (2013) sequenced PITX2c in 100 unrelated patients with lone AF, and found a novel heterozygous PITX2c mutation, p.T97A, in a patient with AF. Functional analysis revealed that the T97A-mutant PITX2c was associated with a significantly decreased transcriptional activity. However, no CHD was observed in these patients with AF-associated PITX2c mutations. Similarly, in mice heterozygous for Pitx2c with left atrial Pitx2c expression being 60% of wild type, the cardiac morphology and function were not altered, but the isolated hearts were susceptible to AF during programmed stimulation (Kirchhof et al., 2011). The pronounced phenotypic variability may be explained by the following reasons. First, AF occurs as rarely as a few times in a lifetime for some patients with AF, suggesting a longer duration of electrocardiographic monitoring required to record paroxysmal AF in our patients. Next, AF occurs more commonly in older patients, and some carriers may not be old enough to develop the disease. Third, considering some congenital cardiac structural defects may close spontaneously, we cannot rule out the possibility that some mutation carriers with lone AF had minor cardiac septal defects that closed shortly after birth on their own. Fourth, the nature of a mutation and its temporal and spatial effects during cardiac development are a potential explanation for this phenomenon (Benson et al., 1999). Fifth, different genetic backgrounds, including possibly common SNPs altering disease susceptibility, contribute to the variable penetrance of the phenotype. Finally, some mutations may be only a genetic risk factor predisposing to a certain disease, rather than a direct cause, and environmental risk factors may be needed for the onset of the disease.
Conclusion
In conclusion, the present study at first associates PITX2c loss-of-function mutations with congenital ventricular septal defect and double outlet right ventricle in humans, which provides novel insight into the molecular mechanism of CHD, and suggests potential implications for early prophylaxis and allele-specific therapy of CHD.
Footnotes
Acknowledgments
The authors are really thankful to the participants for their devotion to the study. This work was supported, in part, by grants from the National Natural Science Fund of China (81070153, 81270161, and 81271927), the Personnel Development Foundation of Shanghai, China (2010019), the Natural Science Fund of Shanghai, China (10ZR1428000), and the Key Program of Basic Research of Shanghai, China (10JC1414002).
Disclosure Statement
No competing financial interests exist.