
Abstract
Despite significant improvement in our understanding of
Introduction
T
T-ALL occurs through the proliferation of malignant T-cell clones, and these clones may be identified by their T-cell receptor (TCR) repertoire, which is specifically expressed on the T-cell surface. In general, T-ALL results from monoclonally expanded malignant T cells that alternatively express TCR αβ or TCR γδ (Chen et al., 2013a). However, patients with T-cell leukemias and lymphomas carrying more than one malignant T-cell clone have been reported (Menin et al., 2003). In some cases, mixed populations can be found, for example, a minor T-cell clone was found at diagnosis in a patient with acute myeloid leukemia (AML), and this minor T-cell subclone progressively increased and became dominant at relapse (Bellido et al., 2000). In addition, secondary T-ALL following AML has been also reported (Liso et al., 1998). Despite significant improvement in our understanding of T-ALL biology and pathogenesis, many questions remain unanswered, and many new ones arise (Kraszewska et al., 2012). For example, little is known about the characteristics of T-ALL cases with two malignant T-cell clones. In this study, we characterized the distribution of the TCRγδ repertoire and the expression of genes related to T-cell activation and response in two T-ALL cases with two malignant TCR Vδ1+ and Vδ2+T-cell clones compared with two T-ALL cases with only one malignant TCR Vα+T-cell clone.
Materials and Methods
Samples
Two newly diagnosed, untreated T-ALL cases that were identified as carrying two malignant Vδ+T-cell clones were selected for this study: case 1 (C1): a 22-year-old male and case 2 (C2): a 4-year-old male. The clinical characteristics of these two cases were similar with high white blood cell counts (more than 100×109/L) and hepatosplenomegaly. The disease was more aggressive and had relatively poor prognosis. Two newly diagnosed, untreated T-ALL cases that were identified as carrying only one malignant Vα+T-cell clone served as control: case 1: a 40-year-old male with a malignant T-ALL clone characterized as having a Vα26Jα16 rearrangement and case 2: a 20-year-old male with a Vα8-4Jα22 malignant T-ALL clone. Peripheral blood mononuclear cells (PBMCs) were collected from patients, and DNA and RNA extraction was performed according to the manufacturer's recommendations. All procedures were conducted according to the guidelines of the Medical Ethics Committee of the Health Bureau of Guangdong Province in China.
Fine-tiling array comparative genomic hybridization and ligation-mediated polymerase chain reaction
Fine-tiling array comparative genomic hybridization (FT-CGH) and ligation-mediated polymerase chain reaction (LM-PCR) were performed as previously described (Dittmann et al., 2012; Chen et al., 2013a). To achieve a high CGH resolution (<1 kb), which is necessary for subsequent in vitro DNA amplification, a custom designed, high-density fine-tiling long oligonucleotide array of 385,000 oligonucleotides 40–60 bp in length was prepared using the Maskless Array Synthesizer (MAS) technology (NimbleGen Systems, Reykjavik, Iceland). This array, covering 24 Mb of genomic regions, was selected using the Human Genome Browser hg18 assembly (University of California, Santa Cruz, CA). The array included TRAD from chromosome 14q11 (Chr14: 21,130–22,130 kb). Neighboring oligonucleotides with an average distance of 63 bp were grouped in 200, 400, and 1000 bp clusters. After normalization with reference DNA (HEK293 T-cell line), the mean fluorescence was analyzed using SignalMap software (NimbleGen Systems). Regions demonstrating DNA loss through FT-CGH were further analyzed by LM-PCR as previously described (Przybylski et al., 2005; Przybylski et al., 2010; Dittmann et al., 2012). The primers used for the LM-PCR were listed in Table 1.
Reverse transcription polymerase chain reaction and GeneScan analysis
RNA extracted from PBMCs from patients with T-ALL was reverse transcribed into cDNA. Primers specific for the TCR Vδ1, Vδ2, and Cδ1 segments (Table 2) were used for PCR amplification. Reverse transcription-polymerase chain reaction (RT-PCR) and GeneScan analysis for the TCRγδ subfamilies were performed as previously described (Geng et al., 2012; Chen et al., 2013a).
Real-time RT-PCR
To compare the expression level of Vδ1 and Vδ2 in the malignant T-cell clones, we used Vδ1-Cδ1 or Vδ2-Cδ1 primer pairs (Table 2) to amplify both gene segments by real-time PCR. PCR was performed as previously described (Geng et al., 2012; Chen et al., 2013a). In addition, to compare the gene expression characteristics of T-ALL cases with Vδ1 and Vδ2 T-cell clones and T-ALL cases with a Vα T-cell clone, the Notch1, TAL1, MALT1, CARMA1, BCL10, A20, and NF-κB mRNA expression levels were detected by real-time RT-PCR using specific primers (Table 2). The relative amount of the genes of interest and the β2M reference gene was measured in two independent assays. Specific amplification of the PCR products was confirmed by melting curve analysis. The data are presented as the relative expression of the genes of interest compared with the internal control gene as determined by the 2(−ΔCT) method (Zha et al., 2012; Shi et al., 2013).
Results and Discussion
The malignant T-cell clones in T-ALL
The combination of FT-CGH and LM-PCR analysis is a novel approach for characterizing novel chromosomal translocations at the molecular level (Przybylski et al., 2005; Przybylski et al., 2010; Dittmann et al., 2012). For lymphocytic malignancies, it is also an accurate technique for identifying malignant T-cell and B-cell clones. In previous studies, we could clearly identify malignant T-cell clones based on analysis of breakpoints at the TCR locus using this technique (Chen et al., 2013a). Moreover, this technique has allowed for the identification of more than one malignant T-cell clone, and we found a case with two malignant T-ALL clones with Vδ1Dδ2Dδ3Jδ1 and Vδ2Dδ3Jδ2 rearrangements (Zheng et al., 2011b). According to the cancer clonal theory, leukemia develops from a single cell due to malignant transformation (Kraszewska et al., 2012). Therefore, every T-ALL clone has a unique TCR gene rearrangement, and T-ALL clones express either TCR αβ or TCR γδ (Chen et al., 2013a). However, few reports have described the characteristics of T-ALL cases containing two malignant clones. In this study, we characterized a T-ALL case with two malignant clones. FT-CGH analysis of the TCR αδ locus (chr14: 21,130–22,130) in the T-ALL sample revealed breakpoints at the 216,300-kb (TCR Vδ1), 219,600-kb (TCR Vδ2), and 219,900-kb (TCR Jδ1) loci (Fig. 1). LM-PCR using nested forward primers specific for the TCR Vδ1 or Vδ2 locus revealed two rearrangements: TCR Vδ1 to Jδ1 and TCR Vδ2 to Jδ1. Direct sequencing of these LM-PCR products demonstrated the following details for the Vδ1Dδ3Jδ1 and Vδ2Dδ1Jδ1 rearrangements (Fig. 1). To verify that these were functional TCR rearrangements, we used RT-PCR to detect the rearrangements with specific primers, and clear PCR products were detected (Fig. 2). This result may indicate that multiple malignant T-cell clones exist in some T-ALL cases, and these can be identified using new techniques such as FT-CGH and LM-PCR. Differences in TCR rearrangements in diagnostic samples may indicate the divergent subclonal evolution of an original preleukemic clone (Zuna et al., 2003). Umino et al. (2011) demonstrated that the genomic profiles of PB from 13 patients with acute adult T-cell leukemia/lymphoma (ATLL) frequently differed from those of lymph node (LN) samples using oligo-array CGH analysis, indicating that multiple subclones in the LNs originate from a common clone and a selected subclone from the LN subclones appears in the PB in many ATLL cases. Moreover, a recent finding showed that more than one-third of late T-ALL recurrences are a second T-ALL incidence that demonstrates different TCR rearrangements and patterns of genomic aberrations (Szczepanski et al., 2011). Although none of the above studies provided direct data demonstrating the identification of two malignant T-cell clones in the same sample at the same time, these findings at least partially support our findings. In this study, two malignant T-cell clones could be identified in the same sample at diagnosis, and this may be related to use of the FT-CGH and LM-PCR techniques, which can display chromosomal breakpoints and directly identify T-cell clones.
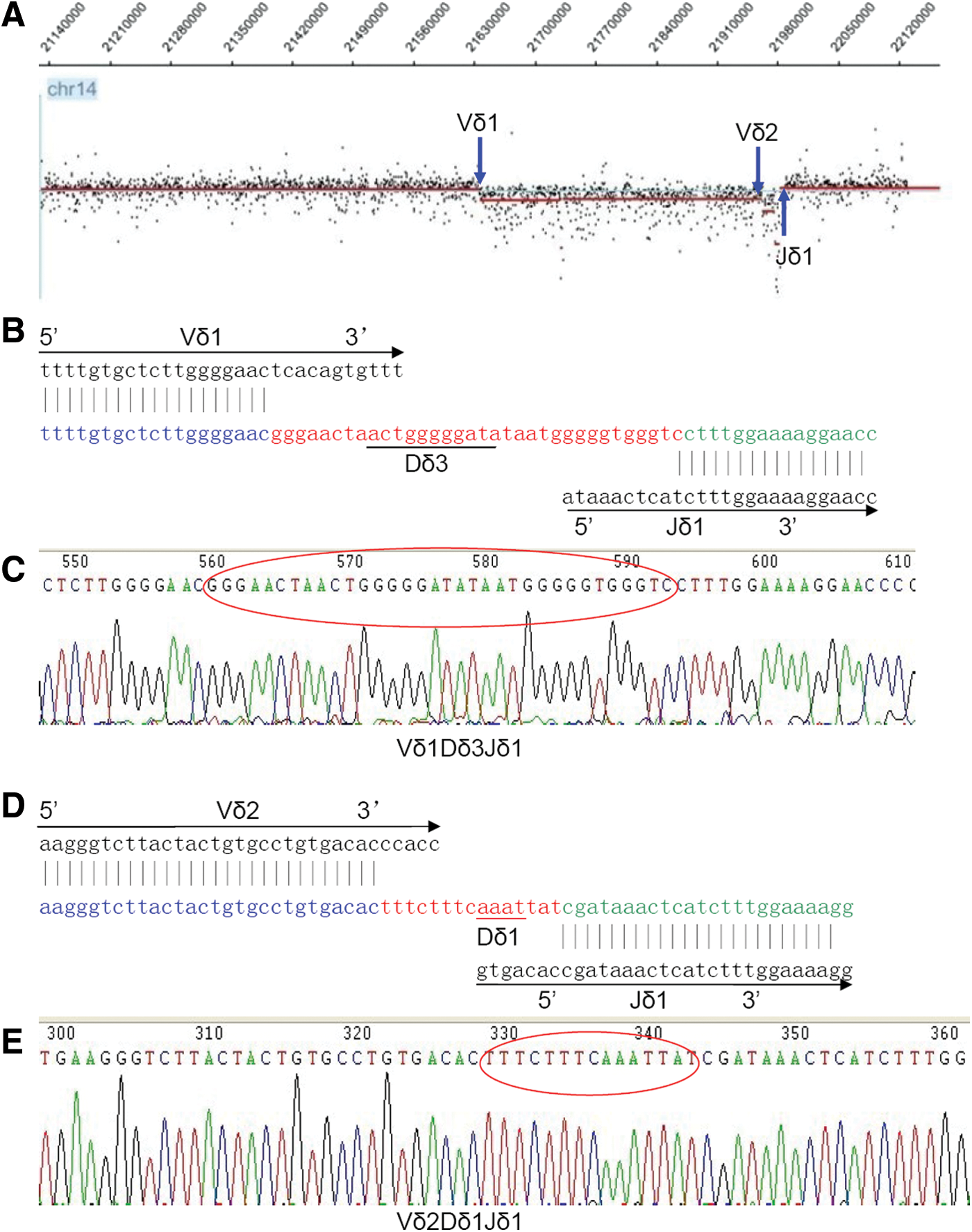
Identification of two malignant T-ALL clones by FT-CGH, ligation-mediated polymerase chain reaction (LM-PCR), and sequencing in a T-ALL case (C1). Vδ1Dδ3Jδ1 and Vδ2Dδ1Jδ1 rearrangements were confirmed.
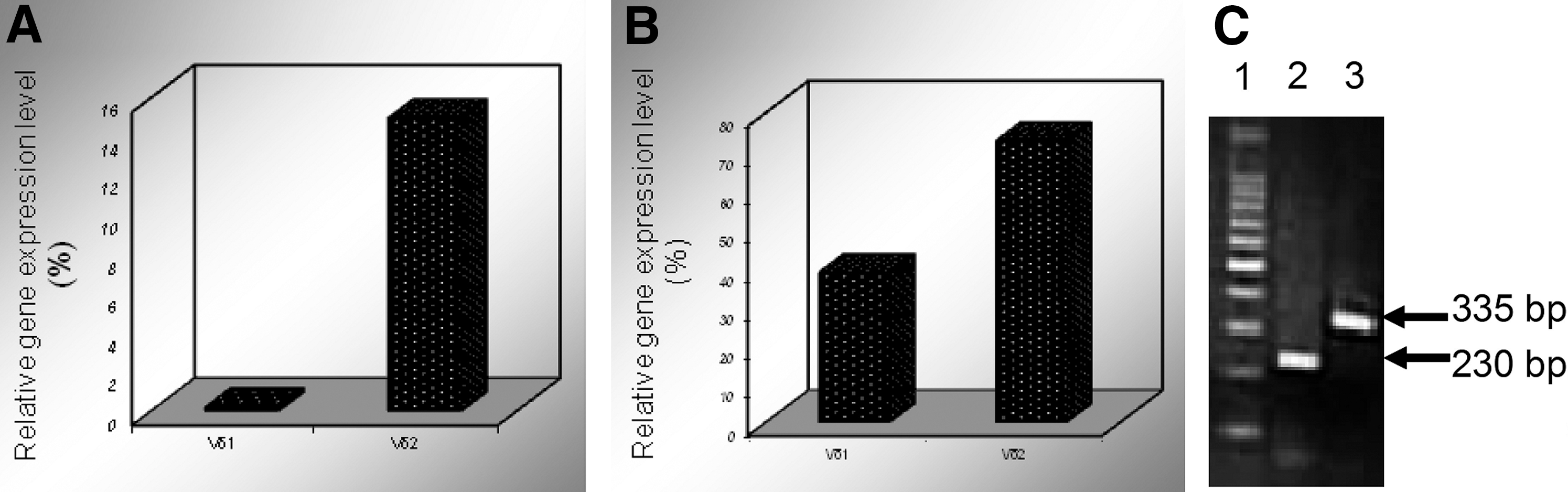
The expression of the Vδ1 and Vδ2 clones in two patients with T-ALL.
Of 14 T-ALL cases (data not shown), we found only 2 cases containing 2 malignant clones. Interestingly, all of the cases expressed Vδ1 and Vδ2 rearrangements. The Vδ2 subfamily is most frequently used in healthy individuals, while the Vδ subfamily members preferentially expressed in T-ALL clones include Vδ1 and Vδ2 and this is similar to our finding (Langerak et al., 1999; Kode et al., 2004). In addition, it may be well understood that the most frequent Vδ T-cell clones may be attacked during T-cell differentiation. We then analyzed the expression level of the Vδ1 and Vδ2 T-cell clones and found that Vδ2 was highly expressed in both T-ALL cases, while the expression of Vδ1 was low, particularly in case 1 (Fig. 2). Thus, this finding may indicate that the Vδ2+T-cell clone is the main malignant T-cell clone in T-ALL patients with two malignant clones, and the Vδ1+T-cell clone may be a minor subclone. However, confirmation by FACS using specific Vδ subfamily antibodies, which can directly show the percentage of malignant T-cell clones, is needed. Thus, these malignant T-cell clones may at least serve as biomarkers for the detection of minimal residual disease (MRD) (Kode et al., 2004; Toubai et al., 2005).
The TCR γδ repertoire in biclonal malignant T-ALL
We further analyzed the distribution and clonality of the TCR Vγ and Vδ subfamily T cells in two patients with T-ALL with two malignant T-cell clones by RT-PCR and GeneScan. Monoclonal Vδ1 and Vδ2 subfamilies were confirmed in both the T-ALL samples, and Vδ3 through Vδ7 could not be detected in the T-ALL samples, whereas the oligoclonal Vδ8 subfamily could be identified in both samples (Fig. 3). These results were further supported by the identification of two malignant Vδ1 and Vδ2 T-cell clones in the T-ALL cases with two malignant clones. However, the oligoclonal expanded Vδ8 T cells were thought of as reactive T-cell clones, which respond to leukemia-associated antigen (Cabillic et al., 2010; Yin et al., 2011), and unlike the malignant T-cell clone, the percentage of this clone was low; thus, the number of T cells containing the same breakpoint in the TCR locus could not be detected by FT-CGH. Therefore, a combination of the FT-CGH and GeneScan techniques may provide an accurate representation of the different properties, clonality, and distribution of TCR subfamilies. This protocol may not only provide biomarkers for MRD, but also facilitate the design of tailored and targeted therapies and immunotherapy for T-ALL subsets (Cabillic et al., 2010; Yin et al., 2011; Wang et al., 2012).
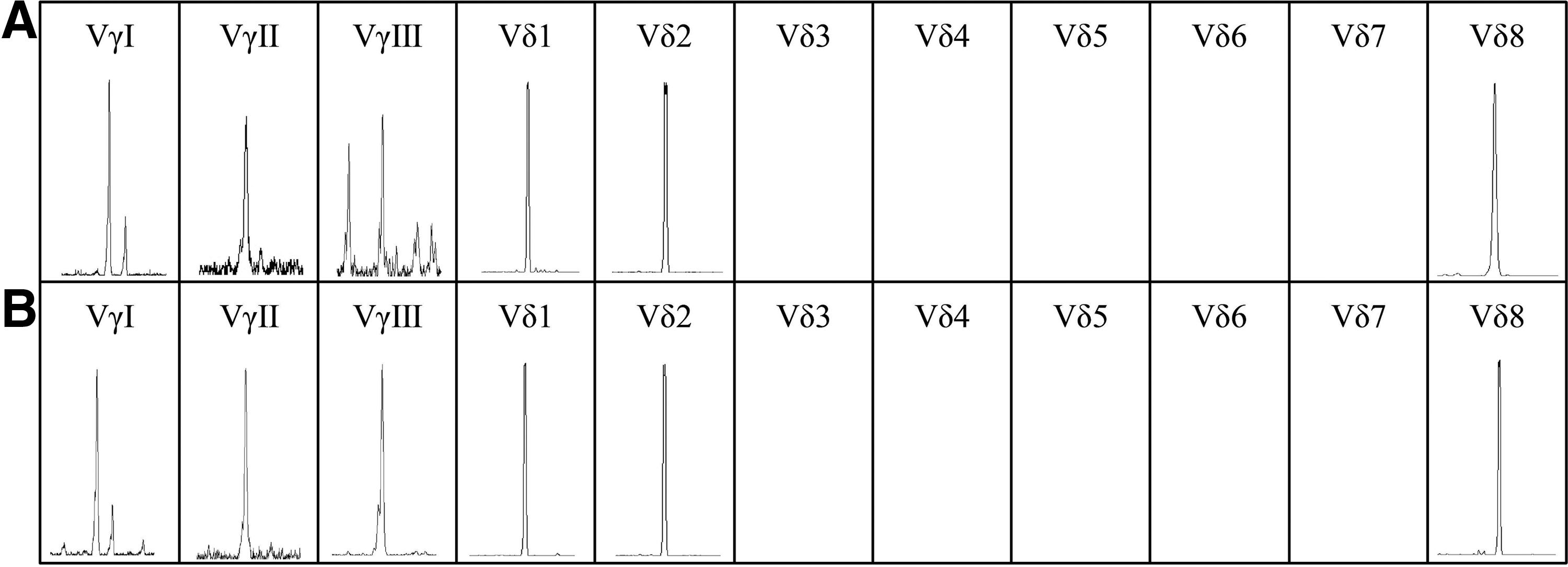
The distribution and clonality of the TCR Vγ and Vδ subfamily T cells in two patients with T-ALL.
The relative gene expression pattern characteristics of T-ALL cases with biclonal malignant Vδ1 and Vδ2 T-cell clones
It has been noted that most T-ALL patients display pathological gene expression even in the absence of chromosome aberrations. Ferrando et al. were the first to report different gene expression patterns in T-ALL using microarray analysis (Ferrando et al., 2002; Kraszewska et al., 2012). Whereas different gene expression patterns may be linked to different prognoses for T-ALL, it is notable that T-ALL patients with Notch1 mutations have different outcomes depending on the therapeutic protocol applied (Kraszewska et al., 2012).
Based on the clinical finding that both the T-ALL cases with bi-malignant T-cell clones had poor outcome, we attempted to compare the expression pattern of genes related to T-cell activation and proliferation between the cases with bi-malignant Vδ1 and Vδ2 T-cell clones and T-ALL cases with a mono-malignant Vα T-cell clone. We selected two T-ALL cases with Vα26Jα16 or Vα8-4Jα22 rearrangements that were identified by FT-CGH and LM-PCR (data not shown) as control. We analyzed the expression level of Notch1, TAL1, and the CARMA-BCL10-MALT-A20-NF-κB pathway genes. The expression level of these genes in biclonal T-ALL was higher than that in monoclonal T-ALL with the exception of the TAL1 gene (Fig. 4), and only A20 had a significant difference (p=0.0354), which may be due to the limited number of samples, although the gene expression tendencies appeared to be unequivocal.
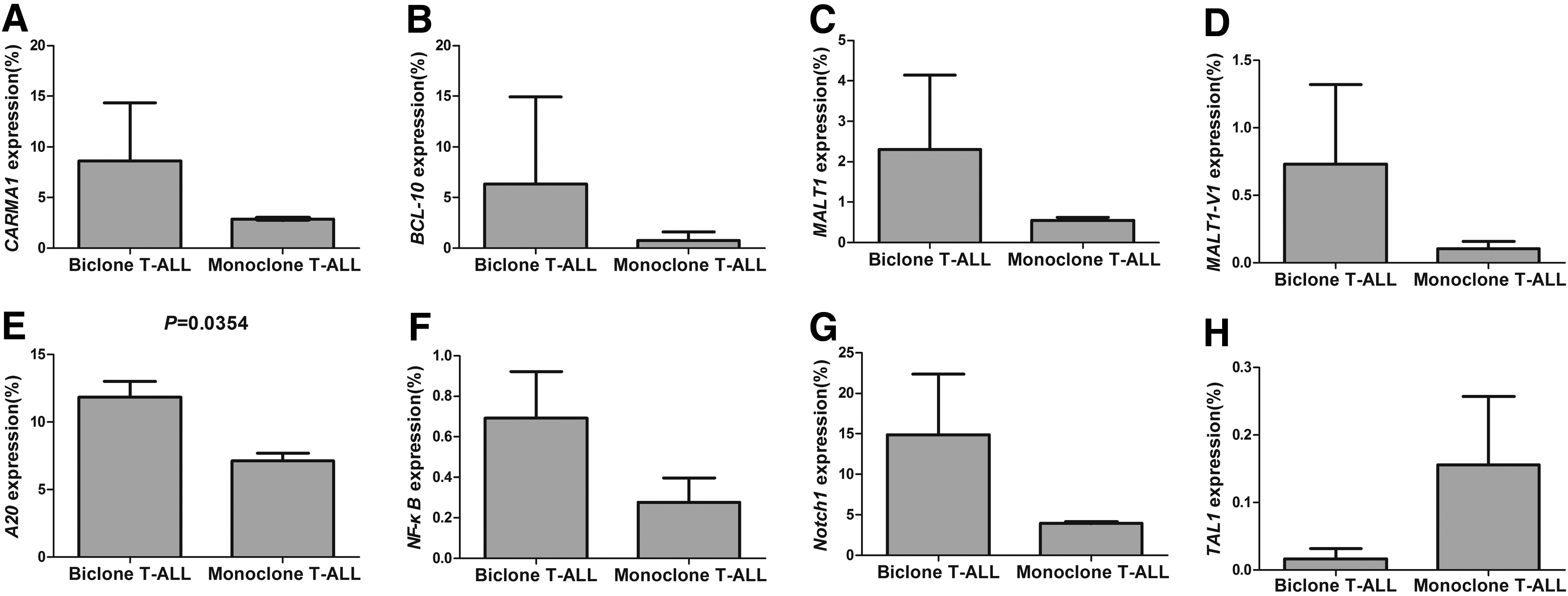
The relative expression level of the CARMA1
Notch1 signaling is crucial for T-cell differentiation and proliferation, and the mutational activation of Notch1 is an important factor in T-ALL pathogenesis (Koch and Radtke, 2011; Zou et al., 2013). Translocation and mutations in Notch1 may alter its function and result in overexpression and independent activation, and ∼60% of T-ALL cases show an increased Notch1 activity (Asnafi et al., 2009; Erbilgin et al., 2010; Koch and Radtke, 2011). The effect of Notch1 activation on the T-ALL outcome is controversial. In this study, a high Notch1 expression level was found in the biclonal T-ALL group, and the significance of its differential expression in biclonal and monoclonal T-ALL requires further investigation. T-cell acute lymphoblastic leukemia 1 (TAL1), also known as stem cell leukemia (SCL), plays an important role in the differentiation of hematopoietic stem cells. Aberrant TAL1 transcription often occurs in T-ALL (Patel et al., 2013). Dysregulation of the TAL1 activity has been associated with T-cell leukemogenesis and initiates T-ALL in mouse models (Tremblay et al., 2010; Li et al., 2012b). Interestingly, a low TAL1 expression level was found in the biclonal T-ALL group, and this result may support the finding that high TAL1 expression is associated with a trend toward good outcome in other T-ALL cases (van Grotel et al., 2008). Because we did not find TAL1 translocations in the four T-ALL cases in both groups (data not shown) using previously published techniques (Bash et al., 1993; Li et al., 2012b; Patel et al., 2013), the differential TAL1 expression pattern requires further investigation.
A20 (also called TNFAIP3) is a zinc finger protein that negatively regulates the expression of proinflammatory genes by downregulating NF-kB activation in response to various stimuli (Beyaert et al., 2000; Coornaert et al., 2008; Zhang et al., 2012). The novel data demonstrating the significant contribution of A20 inactivation resulting in the constitutive activation of the NF-κB pathway is considered to be associated with cancer pathogenesis; A20 deficiency has also been found in the Sezary syndrome, which is a cutaneous T-cell lymphoma, particularly those characterized by constitutive NF-κB activation (Honma et al., 2009; Braun et al., 2012). Our previous findings demonstrated that the A20 expression level was significantly decreased in T-ALL (Ma et al., 2012). Interestingly, the expression level of the A20 gene was significantly upregulated in the biclonal T-ALL group compared with the monoclonal T-ALL group (Fig. 4). A20 is regulated by the CARMA1 (caspase-recruitment domain [CARD]-containing membrane-associated guanylate kinase protein 1, also called CARD11), B-cell lymphoma 10 (BCL10), and mucosa-associated-lymphoid-tissue lymphoma-translocation gene 1 (paracaspase MALT1) (CBM) upstream signaling pathway complex, which bridges TCR signaling with the canonical IκB kinase/NF-κB pathway (Ruland et al., 2003; Coornaert et al., 2008; Li et al., 2012a). We further analyzed the expression levels of the CBM genes, the CARMA1-BCL-10-MALT1-NF-κB expression pattern was high in biclonal T-ALL patients compared with those with monoclonal T-ALL, and this may be one of the gene expression patterns that distinguish both the T-ALL subsets. Few studies have focused on the expression characteristics of the CBM complex in T-cell malignancies. It has been reported that BCL10 is commonly downregulated in peripheral T-cell lymphomas, but has no significant correlation with progression-free survival and overall survival (Rossi et al., 2012). The high expression level of these genes suggests that T-ALL cells have more active proliferation. However, this result is in contrast with the finding that MALT1 mediated the rapid proteolytic cleavage and inactivation of A20 after TCR stimulation (Coornaert et al., 2008; Duwel et al., 2009). Because MALT-1 has two transcripts (MALT1-V1 and V2), we detected not only the total MALT1 expression level, but also the MALT1-V1 expression level (Shi et al., 2013), and also, the latter is expressed higher in the biclonal T-ALL group (Fig. 4). Therefore, whether A20 is linked to negative feedback regulation of NF-kB activation in different T-ALL subtypes remains an open question (Beyaert et al., 2000; Coornaert et al., 2008).
Conclusions
Recent intensive research into the molecular biology of T-ALL revealed significant heterogeneity (Kraszewska et al., 2012). We identified two cases with T-ALL carrying biclonal malignant T-cell clones, and characterized the TCR γδ repertoire and CARMA1, BCL10, MALT1, A20, NF-κB, Notch1, and TAL1 gene expression patterns, which are related to the T-cell activation and proliferation of T-ALL cases with biclonal malignant Vδ1 and Vδ2 T cells. However, the current conclusion was based on the limited two T-ALL cases with biclonal malignant T cells; further data are needed to support the finding by a large cohort analysis. Overall, to our knowledge, this is the first description of such a T-ALL subtype and its gene expression pattern, and this study may improve our understanding of biclonal T-ALL.
Footnotes
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (no. 30871091, 91129720, and 81270604), the Collaborated grant for HK-Macao-TW of the Ministry of Science and Technology (2012DFH30060), the Fundamental Research Funds for the Central Universities (no. 21610603, 21612116), the Guangdong Science and Technology Project (no. 2012B050600023), and Training Programs of Innovation and Entrepreneurship for Undergraduates of Guangdong Province (no. 1055912064).
Authors' Contributions
Y.Q.L. and G.K.P. contributed to the concept development and study design. H.T.Z. performed FT-CGH and LM-PCR analysis, X.W. and Y.M. performed the real-time PCR, S.H.C. performed the RT-PCR and GeneScan, L.J.Y. prepared PBMCs and DNA, X.L.W. and B.L. prepared RNA and cDNA, T.Z.Y., and B.X. were responsible for clinical diagnoses and performed clinical data acquisition. Y.Q.L., H.T.Z., and X.W. coordinated the study and helped draft the manuscript, and S.M.H. and T.Z.Y. helped edit the manuscript. All authors read and approved the final manuscript.
Disclosure Statement
The authors declare that they have no competing interests.