
Abstract
Protein homeostasis in the endoplasmic reticulum (ER) in eukaryotic cells is maintained by a conserved quality control system named ER-associated degradation (ERAD). The ERAD system retains misfolded or unassembled polypeptides in the ER, retrotranslocates them into the cytosol for degradation by the ubiquitin proteasome system. Central to the ERAD process is the AAA+ (ATPase associated with various cellular activities), ATPase p97/VCP (also known as Cdc48p in yeast), and the proteasome. p97/VCP couples ATP hydrolysis to the extraction of misfolded proteins from retrotranslocation sites and subsequently targets them for degradation, but how p97/VCP hands substrate off to the proteasome is unclear. Recent studies suggest that p97/VCP may either directly translocate polypeptides into the proteolytic compartment of the 20S subcomplex, or use a set of shuttling factors to deliver retrotranslocated polypeptides to the proteasome.
Introduction
I
To eliminate polypeptides, the ERAD system employs chaperones to identify and retain folding-defective products. Interestingly, these are the same cohort of enzymes involved in protein folding and assembly, such as BiP, calnexin, and the family of protein disulfide isomerases (Forster et al., 2006; Okuda-Shimizu and Hendershot, 2007; Nakatsukasa and Brodsky, 2008). Thus, while chaperoning polypeptides during the biogenesis process, the same set of factors switch their role at a certain point and turn from a folding catalyst into a degradation triaging factor. The mechanism underlying this decision switch is unknown. However, because the participation of ER chaperones in ERAD is known to involve direct or indirect interactions between substrate-bearing chaperones and protein complexes that serve in the retrotranslocation process (termed retrotranslocon) (Molinari et al., 2002, 2003; Oda et al., 2003; Carvalho et al., 2006; Denic et al., 2006; Gauss et al., 2006; Christianson et al., 2012), it is possible that the interactions of chaperone with retrotranslocon(s), if they are coupled to the relay of polypeptides from the former to the latter, may define an irreversible point beyond which a polypeptide can no longer be brought back to the folding process. Instead, they are now bound to the cytosol for degradation by the ubiquitin proteasome system.
How are misfolded proteins moved across the ER membrane during ERAD? It is generally believed that a few large membrane protein complexes, each assembled around one or more ubiquitin ligases (E3s), may be responsible for retrotranslocation of polypeptides in ERAD (Hampton and Sommer, 2012) (Fig. 1). These complexes may form protein-conducting channels through which polypeptides traverse the lipid bilayer (Carvalho et al., 2010), but how exactly these channels are formed is unknown. What has become clear is that, after crossing the ER membrane, almost every ERAD substrate undergoes polyubiquitination, a posttranslational modification resulting from the attachment of one or more covalently linked ubiquitin chains to a lysine residue (or to a lesser frequency to a serine/threonine site) in substrates (Wang et al., 2007) (Fig. 1). Polyubiquitination is mediated by the sequential actions of three enzymes, a ubiquitin activating-enzyme (E1), a ubiquitin-conjugating enzyme (E2), and an E3 ubiquitin ligase (Ye and Rape, 2009; Christianson and Ye, 2014). As expected, most ubiquitin ligases involved in ERAD either contain membrane anchors or interact with ER-associated proteins, and their catalytic domains all face the cytosol (Hirsch et al., 2009). As a result, ubiquitination occurs on the membrane after a portion of substrate has emerged from the ER lumen, but before the substrate is dislocated into the cytosol. The attached ubiquitin conjugates not only serve as a signal that targets polypeptides to the 26S proteasome for degradation but also facilitate retrotranslocation by engaging a ubiquitin-selective ATPase named p97 or VCP (valosin-containing protein, also known as Cdc48p in yeast) (Bays and Hampton, 2002) (Fig. 1). With a few exceptions, ubiquitinated polypeptides exported from the ER are targeted to the proteasome for degradation. The functional interplays between p97/VCP and the proteasome, which define how substrates are targeted for degradation, are poorly understood. In this review, we discuss the recent progress in understanding how p97/VCP may communicate with the proteasome to deliver ubiquitinated substrates.
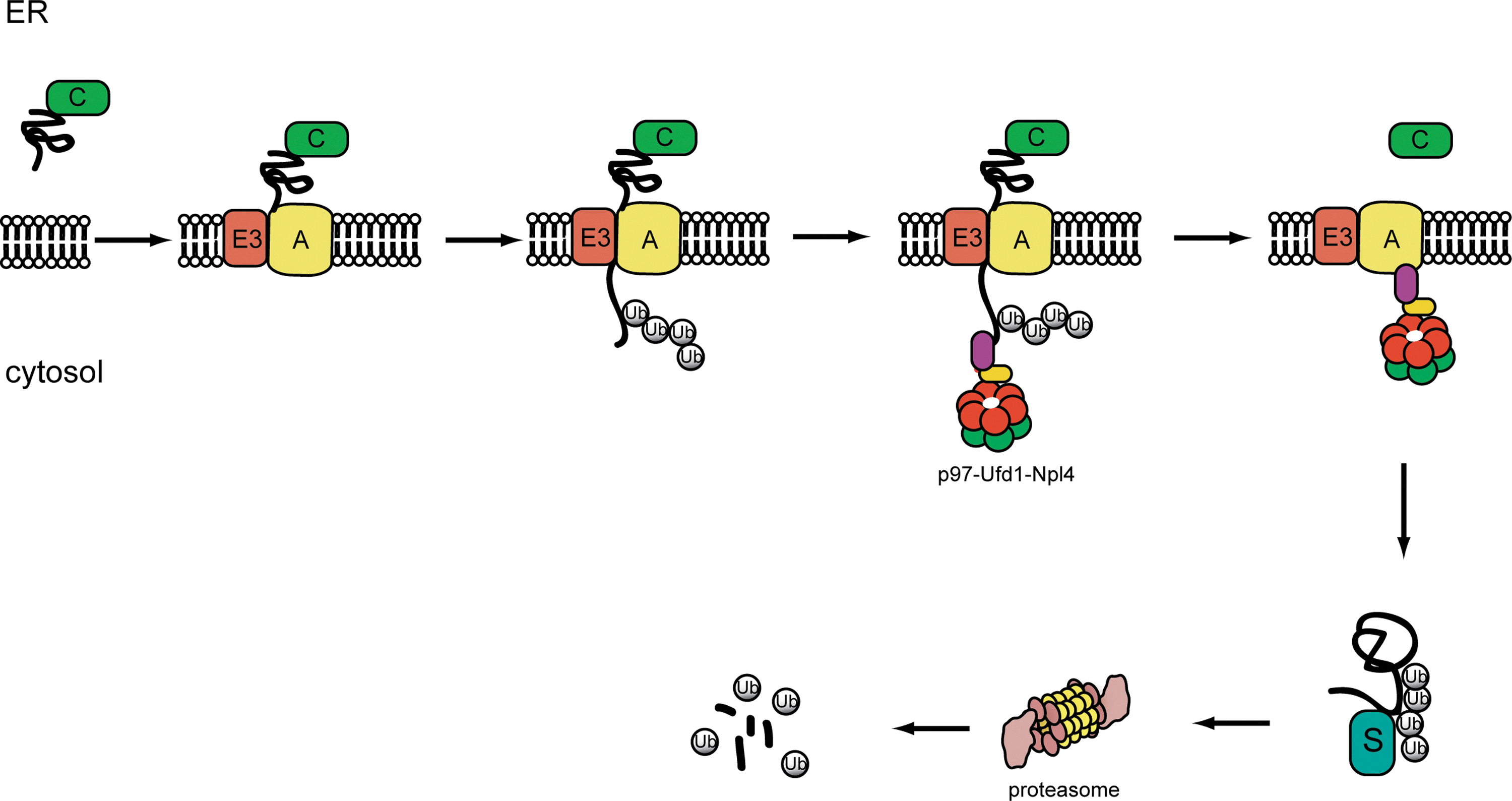
The ER-associated protein degradation pathway. ER-associated protein degradation proceeds through the following steps. In step 1, misfolded proteins are identified by ER chaperones (C), which target them to a retrotranslocation complex in the membrane consisting of E3 ubiquitin ligase and accessory factors (A) (step 2). In step 3, retrotranslocation is initiated as substrate is moved across the ER membrane. The E3 ubiquitin ligase in the retrotranslocation complex adds ubiquitin conjugates to substrate (step 4). Upon modification, the ubiquitinated substrate is pulled out of the membrane by the p97-Ufd1-Npl4 complex. The retrotranslocated substrate is then targeted to the proteasome (step 5) for degradation likely through some shuttling factors (S) (step 6).
p97/VCP: A Multifaceted Cellular Enzyme
Since the discovery that ERAD substrates are degraded in the cytosol, it became apparent that polypeptides need to be retrotranslocated through the lipid bilayer using a process that is likely energy dependent. A search for the enzyme(s) that drives retrotranslocation led to the identification of a cytosolic protein complex consisting of the ATPase p97/VCP and the two cofactors, Ufd1 and Npl4 (Bays et al., 2001; Ye et al., 2001; Jarosch et al., 2002; Rabinovich et al., 2002). p97/VCP forms a homohexameric barrel-like structure that interacts stably with Ufd1 and Npl4 (Pye et al., 2007). This ternary complex contains multiple ubiquitin binding sites that can bind ubiquitinated substrates in a coordinated manner (Ye et al., 2003; Park et al., 2005). The p97/VCP-Ufd1-Npl4 complex plays an evolutionarily conserved role in ERAD as genetic ablation of each component of the complex led to dramatic stabilization of various model ERAD substrates. The stabilized substrates stay mostly in the ER lumen, but a fraction accumulates on the ER membrane in a polyubiquitinated form (Bays et al., 2001; Ye et al., 2001; Jarosch et al., 2002). Thus, the p97/VCP complex is not required for the initiation of retrotranslocation, during which a portion of substrate is pushed out of the ER. Instead, ATP hydrolysis by p97/VCP is coupled to the extraction of substrates from the site of retrotranslocation once the substrates are ubiquitinated (Ye et al., 2001, 2003; Flierman et al., 2003), converting them from a membrane-bound state into a soluble form for degradation.
In addition to ERAD, p97/VCP has been implicated in many other cellular processes such as transcriptional control, fusion of the ER and Golgi membrane, nuclear envelop formation, cell cycle progression, apoptosis regulation, endocytic trafficking, and ribosome-associated degradation (Ye, 2006; Meyer et al., 2012). In each of these pathways, p97/VCP appears to act in conjugation with one or more UBX (ubiquitin regulatory X) domain-containing cofactors to process a distinct class of ubiquitinated substrates, leading to their separation from a large immobile structure (e.g., membranes, chromatins, or sometimes large protein complexes such as the ribosome).
General Action of AAA+ ATPases
p97/VCP belongs to a large ATPase family named AAA+ (ATPase associated with various cellular activities) family. p97/VCP is a type II AAA+ ATPase as it contains two walker-type ATPase domains (D1 and D2). The first ATPase domain is essential for forming the barrel-like hexamer with a central channel (Wang et al., 2004), a common feature shared by many members of this ATPase family. For instance, the six AAA+ ATPases in the base of the 19S proteasome also form a similar ring-like structure. p97/VCP contains an amino-terminal domain (N domain) through which it forms interactions with most cofactors (Schuberth and Buchberger, 2008). In addition, p97/VCP also contains a carboxyl hydrophobic-tyrosine-X motif (HbYX) that was initially identified in the ATPase subunits of the 19S proteasome particle. This motif in the 19S proteasome has been shown to form interactions with the 20S proteasome that control substrate entry into the proteasome (Smith et al., 2007). However, the C-terminus of p97/VCP has also been implicated in association with a few cofactors, including Ufd2/E4b and the cytosolic PNGase (peptide N-glycosidase) (Yeung et al., 2008). Thus, it is unclear whether p97/VCP is able to interact with 20S proteasome in cells.
Two models regarding the mechanisms of AAA+ ATPases have been proposed: the ratcheting model and the substrate threading/unfolding model. In the ratcheting model, an AAA ATPase would grab on a substrate, and conformational changes associated with the ATP hydrolysis cycle may generate mechanical force to separate the substrate from its partners (Halawani and Latterich, 2006). EM studies showed distinct conformations of p97/VCP corresponding to different nucleotide-binding states (Rouiller et al., 2000; Pye et al., 2006). These nucleotide-dependent conformational changes result in a rotational movement of the N-D1 domains relative to the D2 domain, but it is unclear whether such motion is sufficient for force generation. Crystallography studies showed that the N domain of p97/VCP can also move in an up and down motion depending on the nucleotide bound to the D1 domain. If the N domain is immobilized (e.g., on membranes), and the D1 domain moves up and down together with the substrate, this mode of action could operate like a wine opener to separate substrate from the membrane (Tang et al., 2010). One caveat with this model is that purified wild-type p97/VCP always contains ADP in the D1 domain, which does not seem to exchange with ATP efficiently in vitro (Pye et al., 2006). It raises the question of whether the D1 domain ever undergoes efficient ATP hydrolysis in cells.
In the threading/unfolding model, polypeptides are assumed to move through the central pore. This mode of action has been demonstrated for several AAA+ protein machines such as ClpAP, ClpXP, HslU, and Hsp104/ClpB (Halawani and Latterich, 2006). An attractive feature of this model is that substrates threaded through the central channel of an AAA+ ATPase may directly enter the proteolytic chamber of an axially aligned protease oligomer if the latter forms a direct and stable interaction with the ATPase. This mechanism would provide a simple means to couple substrate unfolding to degradation. This is indeed proven to be the case for ClpA and ClpX, both of which interact with the bacterial protease ClpP, and through this interaction, substrates are fed into the ClpP proteolytic compartment by the ClpA or ClpX ATPase (Gottesman et al., 1998; Hinnerwisch et al., 2005). In the case of the disaggregase HSP104/ClpB, polypeptides extracted from aggregates are completely unfolded by threading through the narrow pore of HSP104/ClpB hexamer, which are then passed to other cytosolic chaperones for refolding (Weibezahn et al., 2004). p97/VCP contains ATPase domain architecture similar to these chaperones, and a motif known to interact with the 20S proteasome is also present in p97/VCP. These facts raise the question of whether p97/VCP uses a similar mechanism to extract ubiquitinated polypeptides from the membrane for targeting to the proteasome (see the discussion below).
Transfer Ubiquitinated Polypeptides from p97/VCP to the Proteasome
Direct handoff from p97/VCP to the proteasome?
Once ERAD substrates are extracted from the ER membrane, they should be efficiently targeted for degradation to avoid accumulation of misfolded protein aggregates in the cytosol. This is a critical issue because protein aggregates can sequester cellular factors (e.g., chaperones) to impair essential cellular functions. In this regard, it is not surprising to learn that inhibition of the proteasome activity generally leads to stabilization of ERAD substrates in the ER lumen (Tsai et al., 2002). This observation has long been taken as key evidence in support of a mechanism that couples retrotranslocation to degradation. To explain this coupling phenomenon, it has been postulated that p97/VCP may interact directly or indirectly with the proteasome, which would link retrotranslocation to substrate handoff to the proteasome (Nakatsukasa et al., 2013). In a purely hypothetical model, the hexameric p97/VCP ATPase might be docked directly on the 20S proteasome subcomplex, forming a p97-20S hybrid proteasome in which p97 takes the role of the 19S regulatory particle in regular 26S proteasome (Fig. 2A). Recent studies on the archaeal p97/VCP homolog Cdc48 and the 20S proteasome suggested that direct and functional interactions between Cdc48 and the 20S proteasome can take place in vitro, which align the central cavities of these two ring-shaped complexes, forming a continuous conduit through which substrates are transferred from Cdc48 to the 20S proteasome for degradation (Barthelme and Sauer, 2012, 2013; Forouzan et al., 2012). Such interactions, if they exist in eukaryotes, would provide a simple coupling mechanism that links retrotranslocation to degradation. However, this model is hard to reconcile with biochemical studies showing that p97/VCP does not form stable interactions with 20S proteasomes in eukaryotic cells (Verma et al., 2000; Isakov and Stanhill, 2011), and with genetic evidence showing that both the 19S and 20S proteasome subcomplexes are required for ERAD (Hiller et al., 1996; Jarosch et al., 2002; Christianson et al., 2012). In addition, structural and biochemical studies revealed that the central pore of p97/VCP is blocked at the D1 level, and that only a loop located at the D1 and D2 interface is involved in substrate binding and processing (DeLaBarre and Brunger, 2003, 2005; DeLaBarre et al., 2006). By contrast, the ring formed by the D2 domain contains a wider pore. These observations have prompted the idea that substrates may use the D2 tunnel as an entrance rather than the exit site; Substrates entering the channel formed by the D2 domains may then exist at the interface between D1 and D2 (DeLaBarre et al., 2006). Putting all together, it seems that the threading model may not be a major pathway for transferring substrates from p97/VCP to the proteasome, at least in eukaryotic cells.
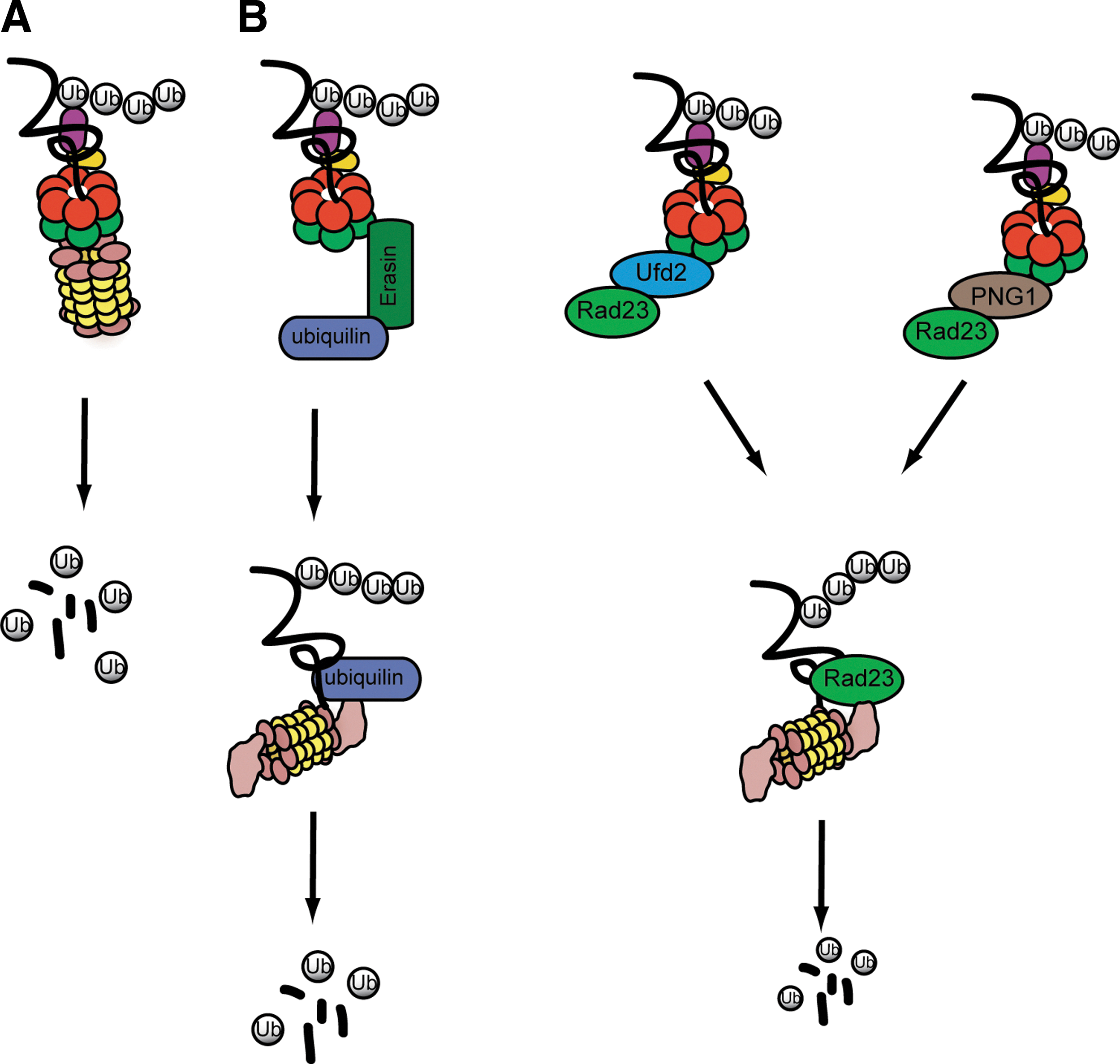
Distinct mechanisms for transferring proteins from p97/VCP to the proteasome.
Transfer substrates using shuttling factors
In contrast to the direct handoff model, the shuttling model proposes that a collection of shuttling factors serve as a mediator to transfer substrates from p97/VCP to the proteasome (Fig. 2B). This model is based on earlier reports that a family of factors containing both ubiquitin-associated (UBA) and ubiquitin-like (UBL) domains can serve as a substrate collector for the 26S proteasome in eukaryotic cells (Kim et al., 2004; Verma et al., 2004), and that in addition to the proteasome, the UBA-UBL–containing factors also interact with the p97/VCP complex through adaptors, which seems to play a role in moving substrates from p97/VCP to the proteasome (Richly et al., 2005). It has been postulated that alternate interactions of shuttling factors with p97/VCP and the proteasome, coupled with substrate loading and unloading, could offer a simple means to shuttle substrates between p97/VCP and the proteasome.
Several p97/VCP adaptors can promote the interaction of UBA-UBL–containing proteins with p97/VCP (Fig. 2B). One is Ufd2p (Richly et al., 2005; Kim et al., 2006). Initially identified in a genetic screen for yeast mutants defective in degradation of a ubiquitin fusion reporter (Johnson et al., 1995), Ufd2p facilitates proteasomal degradation by extending ubiquitin chains formed by a HECT domain E3 ubiquitin ligase (Koegl et al., 1999). Further studies demonstrated that it contains a so-called U-Box domain that shares structural similarity with the RING (Really Interesting Gene) motif commonly found in E3 ubiquitin ligases (Ohi et al., 2003; Tu et al., 2007). Accordingly, it can interact directly with an E2 enzyme to promote transfer of ubiquitin from the E2 to a growing ubiquitin chain in substrates. In cells, Ufd2p can capture ubiquitinated substrates through interaction with the C-terminal tail of Cdc48p (Richly et al., 2005). It also binds the UBA-UBL–containing protein Rad23p through the UBL domain in the latter (Kim et al., 2004). Importantly, the interaction of Rad23p with Ufd2p and the proteasome was found to be mutually exclusive, suggesting that Rad23p probably interacts with Ufd2p and the proteasome in an alternate manner in cells (Richly et al., 2005). On the other hand, a ternary complex containing Cdc48p, Ufd2p, and Rad23p could be detected (Richly et al., 2005). Collectively, these observations indicate that Ufd2p may recruit Rad23p to Cdc48p to facilitate the transfer of substrates from Cdc48p to Rad23p.
It is unclear whether the mammalian Ufd2p homolog E4B has a role in ERAD, but a protein named erasin appears to function similarly to Ufd2p in this process. Erasin is an ER stress-induced protein anchored to the ER membrane. It contains a UBX domain that binds p97/VCP. It also interacts with a UBA-UBL–containing protein named ubiquilin to recruit it to p97/VCP. Knockdown of erasin or ubiquilin inhibits the degradation of several ERAD substrates and induces ER stress, suggesting that these proteins are functionally required for ERAD (Lim et al., 2009). Ufd2p/E4B and erasin may form parallel routes through which distinct substrates are flowed from p97/VCP to the proteasome using different UBA-UBL–containing proteins. In addition, the cytosolic PNGase (peptide N-glycosidase) may constitute another pathway dedicated to targeting of misfolded glycoproteins in yeast and mammals (Hirsch et al., 2003; Kim et al., 2006).
In addition to the above-mentioned p97/VCP adaptors, recent studies also suggested that a specific chaperone system may cooperate with UBA-UBL–containing factors during this process in mammalian cells. The system comprised the Bag6–Ubl4A–Trc35 complex and the cochaperone SGTA (Lee and Ye, 2013). The central player, Bag6, is an abundant cytosolic chaperone with a fraction of it associated with the ER membrane. The membrane interaction of Bag6 is mediated through interactions of its N-terminal UBL domain with gp78 and UbxD8, two components of a retrotranslocation complex (Christianson et al., 2012; Xu et al., 2013). Bag6 contains a large proline-rich segment predicted to be unstructured. Nonetheless, the proline-rich domain is sufficient to form an oligomer that binds substrates bearing long hydrophobic segments (Xu et al., 2013). This interaction can maintain a misfolded polypeptide in an unfolded yet soluble state (Wang et al., 2011). In cells, Bag6 preferentially captures dislocated ERAD substrates downstream of p97 (Claessen and Ploegh, 2011; Wang et al., 2011). This chaperone activity appears to facilitate the degradation of some ERAD substrates by preventing their aggregation (Wang et al., 2011). Thus, the most plausible interpretation of these results is that Bag6 acts between p97/VCP and the proteasome to channel substrates from the sites of retrotranslocation to the proteasome. In agreement with this idea, Bag6 was also found to interact with the proteasome and ubiquilin (Xu et al., 2012), but whether these interactions are direct or simply a result of all factors binding to the same substrate as it is being targeted to the proteasome is unclear. It is also worth mentioning that although many aspects of the ERAD system are conserved in Saccharomyces cerevisiae, the budding yeast does not contain a Bag6 homolog. A homologous system consisting of Sgt2p, Get5p, and Get4p (homologs of SGTA, Ubl4A, and Trc35, respectively) was found in yeast, but it is mainly involved in targeting tail-anchored proteins to the ER membrane (Lee and Ye, 2013). However, a group of small cytosolic heat-shock proteins may serve an analogous function, as suggested by a previous study (Ahner et al., 2007).
Perspectives
We have now come to a point that most (if not all) components involved in shuttling dislocated ERAD substrates from p97 to the proteasome have been identified, but how these factors act together to fulfill this important task is unclear. Many questions are to be addressed by future studies. Among them, the most important one is regarding the mechanism by which p97/VCP dislocates ERAD substrates from the membrane. It is important to understand whether or not the p97/VCP ATPase threads substrates through its central pore during retrotranslocation. To answer this question, we need a quantitative in vitro assay, in which recombinant p97/VCP is able to extract ubiquitinated model substrates from either vesicles or a preassembled protein complex. Assays of this kind would allow implementing structure-based mutagenesis studies to address the question of whether substrates ever enter the central cavity of p97/VCP. Another relevant question is whether or not p97/VCP ever forms stable interactions with either the 26S proteasome or the 20S subcomplex in cells under certain circumstances, allowing direct coupling of retrotranslocation to degradation. Addressing these questions would help to determine whether the direct handoff pathway exists in eukaryotic cells, and if so, whether this model or the shuttling pathway represents the major mechanism for degradation of p97/VCP substrates. For the shuttling pathway, many parts of the outlined model remain at a speculative stage, and thus require experimental proofs. Whether the UBA-UBL–containing factors act in parallel pathways or in a hierarchy system is another important question. Answers to these questions will likely impact research in many other processes that involve p97/VCP given the broad partnership between p97/VCP and the proteasome. However, studies in this field have been hindered by the presence of multiple homologous proteins in every eukaryotic species for many involved factors. For example, the human genome encodes four ubiquilin homologs. As a result, the conventional genetic approach often fails to reveal the function of these proteins because of potential redundancy. Fortunately, the recently developed genome editing technologies have made it possible to knock out several genes simultaneously. The technological advancement will for sure provide an exciting platform for biologists to tackle these long-standing questions.
Footnotes
Acknowledgments
The authors' research is supported by the NIH intramural AIDS Targeted Antiviral Program (IATAP) and by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Disclosure Statement
No competing financial interests exist.