
Abstract
Few studies have referred to the implication of anoikis processes following hormonal treatment. No data are available on the influence of estrogen in ovarian cancer anoikis. To gain insights into the effects and mechanism of estrogen in ovarian cancer cells, we have carried out studies on the anoikis of ovarian cancer cells treated with estrogen and on the pathways involved. We observed an anti-anoikis role of E2 in suspended Caov-3 cells, and this was mainly due to the decreasing of Bit1 level in cytosol. We also found that estrogen receptor α (ERα) was the main mediator involved in this process. To study the signaling pathways well, phosphatidylinositol 3-kinase (PI3K)/AKT were further investigated. Results demonstrated that the decreasing of the Bit1 level in cytosol mediated by E2 binding to ERα was mainly through PI3K/AKT pathways. Overall, these findings disclose a new perspective for estrogen on ovarian cancer therapy.
Introduction
O
Estrogens are a class of steroid hormones secreted by granulosa cells in the ovary, which are essential for follicle development and fertility. They are decisive to the functioning and maintenance of a diverse array of tissues and physiological systems in mammals. The roles of estrogen on such classical targets as the reproductive tract, gonads, mammary tissue, and hypothalamic/pituitary axis have been well identified. A part in nonreproductive tissues, such as maintenance of bone mineral density and cardiovascular health in women, also has been illustrated (Miller et al., 1995; Bush et al., 1996). Estrogens occur naturally in several structurally related forms, but the predominant intracellular estrogen is 17β-estradiol (E2). The physiological responses to estrogen are known to be mediated by at least two estrogen receptors (ERs), ERα and ERβ. ER gene expression has been demonstrated to be restricted to specific tissues and under complex hormonal control. The ERs are a class I member of the nuclear hormone receptor family and act as ligand-activated nuclear transcription factors. Studies of the receptors' tissue distribution and expression pattern showed that ERα has a broad expression pattern, whereas ERβ has a more focused pattern with high expression in the ovary, prostate, epididymis, lung, and hypothalamus (Couse et al., 1997; Kuiper et al., 1997).
There is evidence that estrogens promote tumor development and progression in EOCs (Sasaki et al., 2008; Mǿrch et al., 2009). Primary cultures of human ovarian cancer cells secrete 17β estradiol (E2) (Wimalasena et al., 1991) and estrogen has been shown to provoke the proliferation of human ovarian surface epithelium and ovarian cancer cells (Syed et al., 2001). Epidemiological studies implicate that estrogen treatment may be responsible for initiating ovarian cancer development in postmenopausal women (Rodriguez et al., 1995; Gambacciani et al., 2003). Microarray analyses of ovarian cancer cells treated with estrogen suggest that expression of the cancer-related genes cyclin B1, tumor necrosis factor type 1 receptor (TRAP1), AP4 DNA binding protein (TFAP4), lipocalin 2, hypoxia-induced factor-1a (HIF-1a), vascular endothelial growth factor-A (VEGF-A), and fibulin-1C (Moll et al., 2002; O'Donnell et al., 2005) are increased. These genes activate the signaling pathways associated with ovarian cancer such as angiogenesis. For example, HIF-1a, VEGF-A, and GPR30 activate PI3/Akt in response to estrogen in ovarian cancer cells (Kimura et al., 2004; Revankar et al., 2005). However, there are opposite studies (Santen et al., 2010) supporting the use of estrogens for women with debilitating menopausal symptoms, with reservations in only a small subset of cancers. In brief, the precise relationship of estrogens and ovarian cancer is still open for debate.
The survival of adherent cells is anchorage dependent. Loss of attachment causes cell death through an apoptosis process known as anoikis (Frisch and Ruoslahti, 1997). Tumor cells tend to be less dependent on attachment to the extracellular matrix (ECM) and more resistant to anoikis than normal cells. This resistance to anoikis enables tumor cells to survive the lack of attachment during invasion and metastasis, and this process is essential for the development of tumor. The signaling pathways that prevent anoikis originate from integrin-mediated attachment of cells to the ECM, and some of the well-known integrin signaling molecules, such as focal adhesion kinase, have been illustrated to regulate anoikis (Frisch et al., 1996; Schwartz, 2001).
Bit1 (Bcl2-inhibitor of transcription 1) is a mitochondrial protein evolutionarily conserved from bacteria to humans (Jan et al., 2004). Compared to bacteria Bit1, the 22-kDa Bit1 polypeptide of archaea and eukaryotes contains an extra N-terminal mitochondrial localization domain, which localize Bit1 to the outer mitochondrial membrane. The role of Bit1 serving as one of the two enzymes that release tRNA from a peptidyl-tRNA complex has been well illustrated. Hence, the protein also is called peptidyl-tRNA hydrolase 2. As a mitochondrial protein, Bit1 has another function: It becomes part of the machinery that brings about cell death upon its release from mitochondria into the cytosol or when experimentally expressed in cytosol where it forms a complex with AES (a member of the Groucho family of transcriptional corepressors) and promotes apoptosis (Jan et al., 2004). Various antiapoptotic signaling molecules, such as Bcl-2, Bcl-xL, phosphatidylinositol 3-kinase (PI3K), and AKT, can interdict the release of Bit1 from mitochondria, but are incapable to inhibit apoptosis induced by cytosolic Bit1. Only integrin-mediated cell attachment counteracts cytosolic Bit1. Thus, this caspase-independent apoptosis pathway seems to be particularly important in anoikis.
Anoikis plays an important role in the metastasis and invasion of ovarian cancer, which has been associated with incidence of ovarian cancer. However, the influence of estrogens on anoikis of ovarian cells was barely investigated. Although studies have shown that ER plays a critical role in anoikis and invasion of squamous cell carcinoma (Ishida H et al., 2007). In this study, we set out to test the hypothesis that estrogens may have influence on the anoikis process of ovarian cancer cells, and Bit1 may involve in this process. Our results showed that E2 decreases Bit1 level in cytosol, which in turn reduced anoikis of suspended Caov-3 cells, and the effect of E2 was mediated by ERα. We also found that PI3K/AKT was involved in this process. Overall, E2 treatment could impair anoikis of cultured Caov-3 cells at least through reducing cytoplasmic Bit1, which was mainly dependent on PI3K/AKT.
Materials and Methods
Proteins, reagents, and antibodies
Human ovarian papillary adenocarcinoma cell line (Caov-3) cells were obtained from the American Type Culture Collection (Rockville, MD); Bit 1 polyclonal antibody was purchased from the Cell Signaling Technology (Beverly, MA); PolyHEMA (polyhydroxyethylmethacrylate) were obtained from Sigma Chemical Co. (St. Louis, MO.); the BSA protein assay kit, human Bit1 primary antibody and second antibody, and PI3K/AKT inhibitors were purchased from Pierce (Rockford, IL); electrochemiluminescence (ECL) western blotting detection reagents and TRIzol were purchased from TaKaRa (Dalian, China); the Annexin V and propidium iodide (PI) double staining kit and 17b-estradiol were purchased from Sigma (St. Louis, MO); the protein assay reagent were obtained from Bio-Rad Laboratories (Richmond, CA); Lipofectamine 2000™ transfection reagent, small interfering RNA (siRNA) against ERα and ERβ, Dulbecco's modified Eagle's medium (DMEM), and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, NM); All other products were obtained from Sigma-Aldrich (St. Louis, MO).
Cell culture
Human ovarian papillary adenocarcinoma cell line Caov-3 cells were cultured in DMEM containing 10% FBS, 100 U/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate. Cells were maintained at 37°C in a 5% CO2-enriched humidified air atmosphere.
Suspended cell culture
Caov-3 cells were cultured and harvested by typsine at 80% confluence. Five milliliters of suspended cell cultures (containing 5×104 cells/mL) were seeded on 60-mm polyHEMA dishes and maintained at 37°C in a 5% CO2-enriched humidified air atmosphere. PolyHEMA dish was made as previously described (Zhao et al., 2011): 2 mL polyHEMA (10 mg/mL polyhydroxyethylmethacrylate, diluted in ethyl alcohol) were plated in the dish twice and allowed to dry on a clean bench for 24 h. Before it was used, the dish was washed with sterile phosphate buffered saline (PBS) three times.
Estradiol treatment
Suspended cell cultures were prepared as above. Briefly, 5×104 cells were seeded in 60-mm plates coated with 10 mg/mL polyHEMA in a regular medium. For subsequent experiment, 100 nM E2 was added to the cell culture for 24 h. For control, no E2 was added to cell cultures.
Analysis of apoptosis
Apoptosis was examined by flow cytometry with the Annexin V-FITC/PI double staining kit according to the manufacturer's instruction. Briefly, suspended Caov-3 cells treated with or without E2 were harvested and washed with cold PBS. 5×105 cells were resuspended in 100 μL of 1× Annexin-binding buffer and incubated with 5 μL of FITC-labeled Annexin V and 1 μL of 100 μg/mL PI working solution at room temperature for 20 min, followed by the addition of 400 μL of 1× Annexin-binding buffer, mixed gently, and kept on ice. After staining, apoptotic cells showed green fluorescence (Annexin V-FITC-positive), dead cells showed red and green fluorescence, and live cells showed little or no fluorescence signal. Percent apoptosis was determined with related software.
Western blotting
Mitochondrial and cytoplasmic extracts were prepared from Caov-3 cells using the Mitochondria/Cytosol Fractionation Kit according to the manufacturer's protocol. Total protein concentration in extracts was examined by the Bradford assay using the protein assay reagent according to the manufacturer's protocol. For western blot analysis, cytoplasm, mitochondrial, and total cell lysates were size-fractionated by SDS-PAGE and transferred onto nitrocellulose membranes. Then membranes were blocked with 5% nonfat milk for 1 h. Polyclonal human Bit1 primary antibodies were incubated overnight at 4°C, and Bit1 secondary antibodies were incubated overnight at room temperature (RT) followed by peroxidase-conjugated goat anti-mouse IgG for 1 h at RT. Immunoreactive proteins were visualized with the ECL western blotting detection reagents.
RNA isolation and real-time polymerase chain reaction
Total cellular RNA was isolated using the TRIzol reagent according to the manufacturer's instruction. First strand cDNA was prepared from 1 μg of total cellular RNA in 20 μL of reaction volume using 20 U of M-MuLV reverse transcriptase. Primes were as follows: β-actin sense: 5′-AGA AGG ATT CCT ATG TGG GGG-3′, Anti-sense:5′-CAT GTC GTC CCA GTT GGT GAC-3′; ERα sense: 5′-TGT GCA ATG ACT ATG CTT CA-3′, anti-sense: 5′-GCT CTT CCT CCT GTT TTT A-3′; ERβ sense: 5′-GTC CAT CGC CAG TTA TCA CAT C-3′, anti-sense: 5′-GCC TTA CAT CCT TCA CAC GA-3′. Amplification was conducted using the polymerase chain reaction (PCR) core kit. PCR was performed under the following conditions: 94°C for 4 min, followed by 40 cycles at 94°C for 30 s, 50°C for 30 s, and 72°C for 40 s. Each sample was run in triplicate.
siRNA interference
siRNA against estrogen receptor-ERα/ERβ or Bit1 were used to knock down ERα/ERβ or Bit1 expression in Caov-3 cells. For transient transfection experiments, 2×105 Caov-3 cells were transfected with 25 μM of ERα- or ERβ-specific siRNAs using the Lipofectamine 2000 transfection reagent according to the manufacturer's protocol. ERs and Bit1 gene silencing was confirmed 72 h post-transfection by semi-quantitative reverse transcription-PCR analysis. Total RNA was isolated with the TRIzol reagent. ERs knockdown Caov-3 cells were used to perform E2 treatment, western blotting, and cell apoptosis analyses.
Inhibit assay of PI3K/AKT or p38/MAPK
Caov-3 Cells were treated with PI3K or AKT inhibitors 0.5 h before E2 treatment. Briefly, 10 μM of selective p38 inhibitors LY 2228820 (Selleckchem, Houston, TX) or 10 μM of selective AKT inhibitors CCT128930 (Selleckchem) were added to suspended Caov-3 cell cultures. 0.5 h later, E2 were added to cell cultures for another 24 h incubation, and then cells were collected for western blotting and cell apoptosis analyses.
Statistical analysis
Data are presented as means±SE. All assays were performed at least twice with duplicate or triplicate samples in each experiment. Data were analyzed for statistical significance using a paired Student's t-test. A p-value of <0.05 was considered significant.
Results
E2 treatment reduces anoikis of Caov-3 cells
Although previous investigations have demonstrated that estrogen can induce the proliferation of human ovarian cancer cells, little is known about the effect of E2 on anoikis of ovarian cancer cells. To well understand the effect of E2 on anoikis, we treated suspended Caov-3 cells with 100 nM E2 for 24 h. Cell apoptosis was detected by the Annexin V-FITC/PI staining. Results showed that treating with E2 (Fig. 1B) reduced the apoptosis of suspended Caov-3 cells, to a level that had significant difference with cells at resting (Fig. 1A). In other words, E2 treatment reduced anoikis of Caov-3 cells.
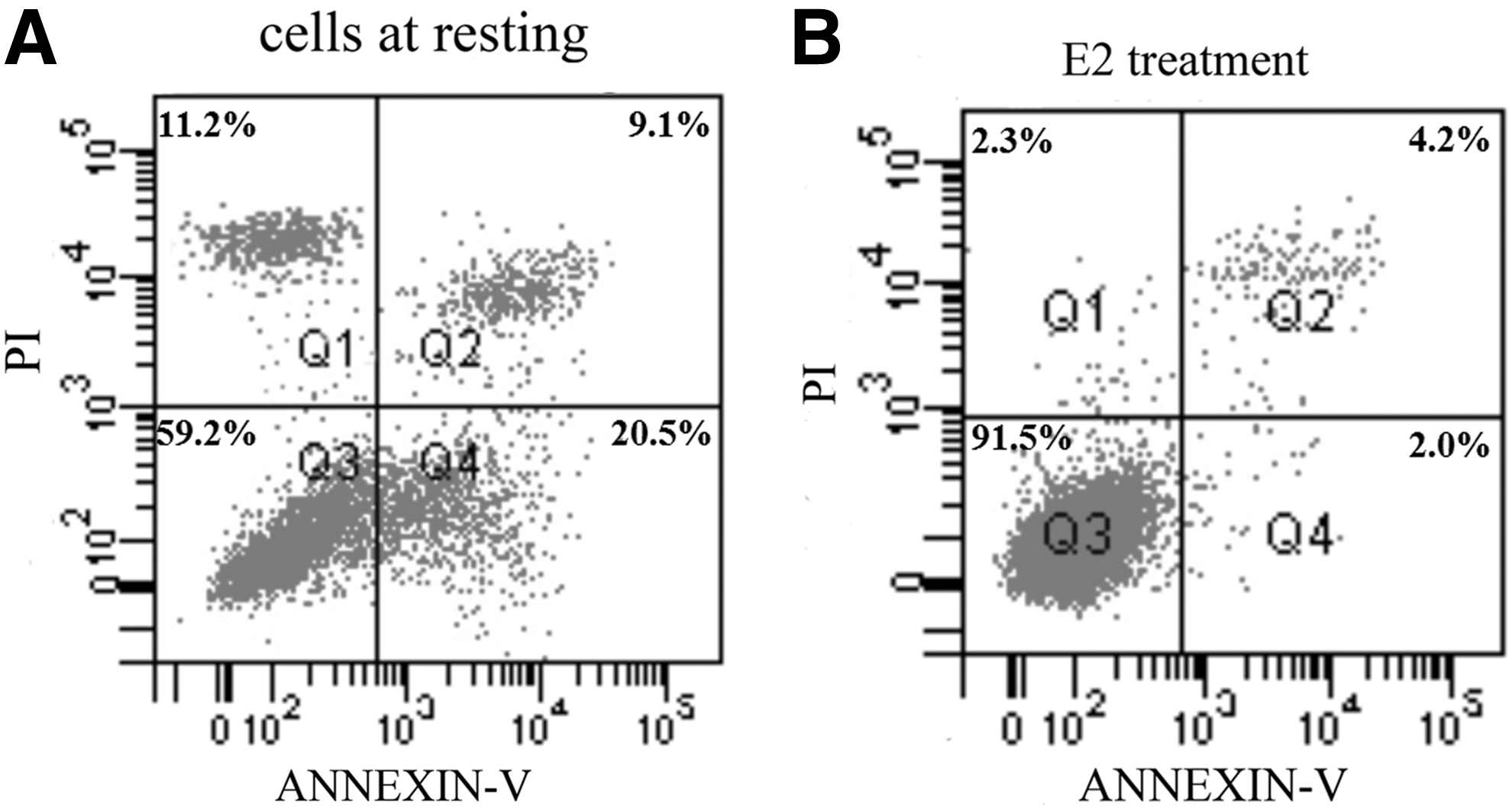
E2 treatment reduces Caov-3 cell anoikis. Suspended Caov-3 cell apoptosis was treated with or without E2, and Annexin V-FITC/propidium iodide was used to determine cell apoptosis.
Determination of Bit1 level in cytosol and mitochondria
As we know, one of the Bit1 functions is acting as a proapoptotic factor when released into the cytosol or when experimentally expressed there. Increasing Bit1 expression in cytosol enhances anoikis (Jan et al., 2004). To confirm that, Bit1-specific siRNA were transfected into Caov-3 cells and cell anoikis were detected (Fig. 2A). As shown in Figure 2B, transfection with Bit1-specific siRNA effectively prevented cell anoikis compared with cells at resting and cells transfected with scrambled siRNA. We hypothesized that Bit1 may be involved in E2-induced decreasing of Caov-3 anoikis. So we detected the Bit1 level change in these processes. Western blotting results showed that Bit1 level in the cytosol of cells treated with E2 was lower compared with control (Fig. 2C). Furthermore, we have analyzed the mitochondrial and total Bit1 levels. The data demonstrated that treatment with E2, the total Bit1 level of cells was neither promoted nor retarded, but the cytosol level decreased and mitochondrial level increased (Fig. 2D). We also performed experiments on the cells overexpression cytosol Bit1, and found that in these cells, E2 treatment could not reduce cell anoikis (Fig. 2E). All these findings suggested that E2 had no influence on Bit1 expression, but could inhibit the release of Bit1 from mitochondria to cytosol, which subsequently could reduce anoikis.
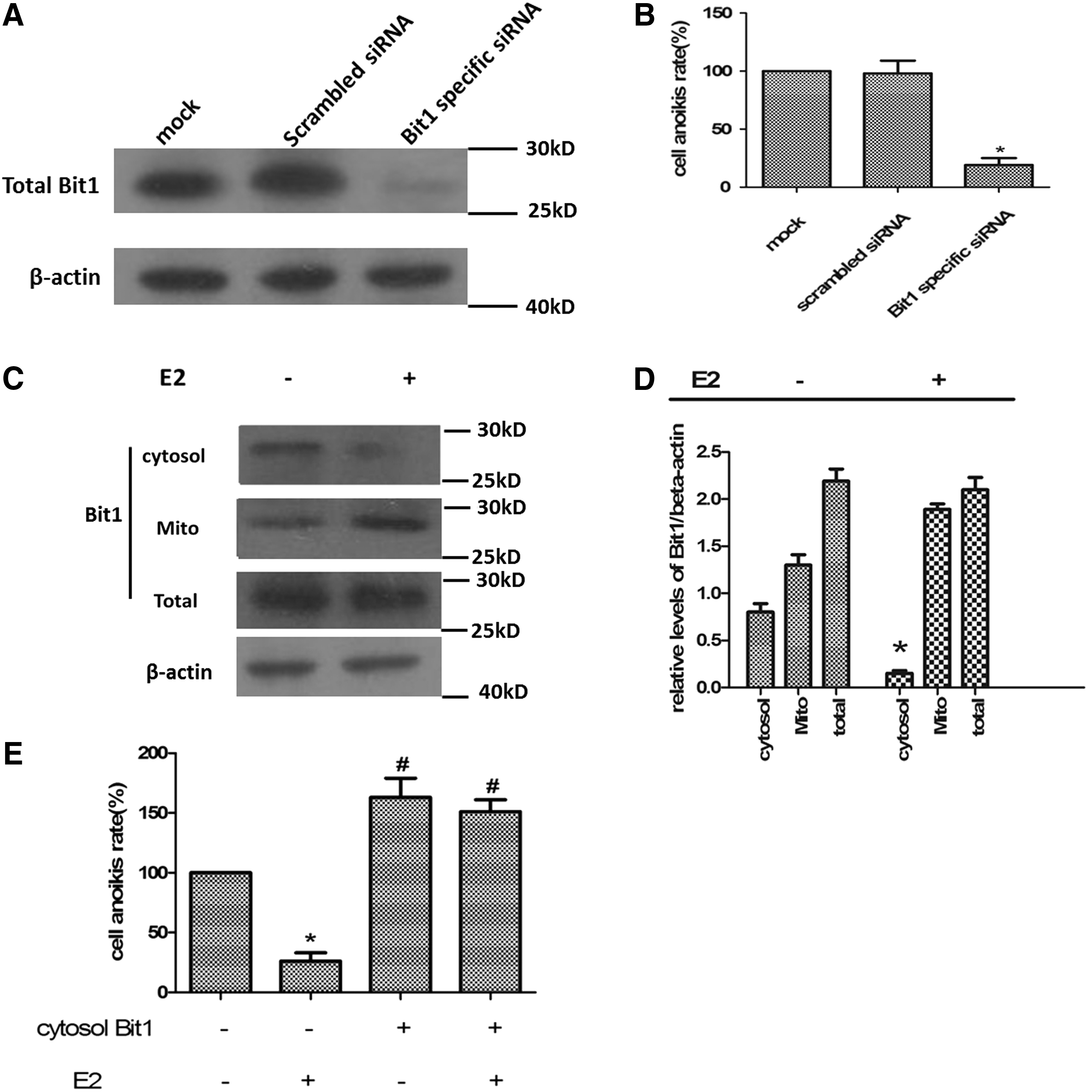
E2 treatment could inhibit the release of mitochondrial Bit1 into cytosol.
E2-induced reduction of anoikis in the presence of ERα
Estrogen effects are mainly mediated by ERα and ERβ. In normal ovaries, ERβ mRNA was the predominant ER form, whereas in ovarian cancer cell lines ERα mRNA was markedly increased as compared with ERβ mRNA (Pujol et al., 1998). So which one played a predominant role in E2-induced reduction of anoikis? To further analyze that siRNAi was adopted to construct ERα or ERβ knockout cell models. Using siRNA, we were able to selectively reduce ERα or ERβ expression to undetectable levels in Caov-3 cells (Fig. 3A). Then we found that in ERα knockdown Caov-3 cells, E2 failed to induce reduction of Bit1 level in cytosol (Fig. 3B). There was no significant difference between ERα knockdown Caov-3 cells and cells at resting (without RNAi and E2 treatment). In ERα knockdown Caov-3 cells, E2 treatment barely inhibited cell anoikis compared with cells treated with E2 only (Fig. 3C). But in ERβ knockdown cells treated with E2, Bit1 level in cytosol reduced to a level which had no difference with the control (E2 treated group without RNAi). Whereas in ERβ knockdown cells, cell anoikis were significantly reduced by E2 treatment (Fig. 3D). These results suggested that it was ERα that mediated the anoikis decreasing function of E2, but not ERβ.
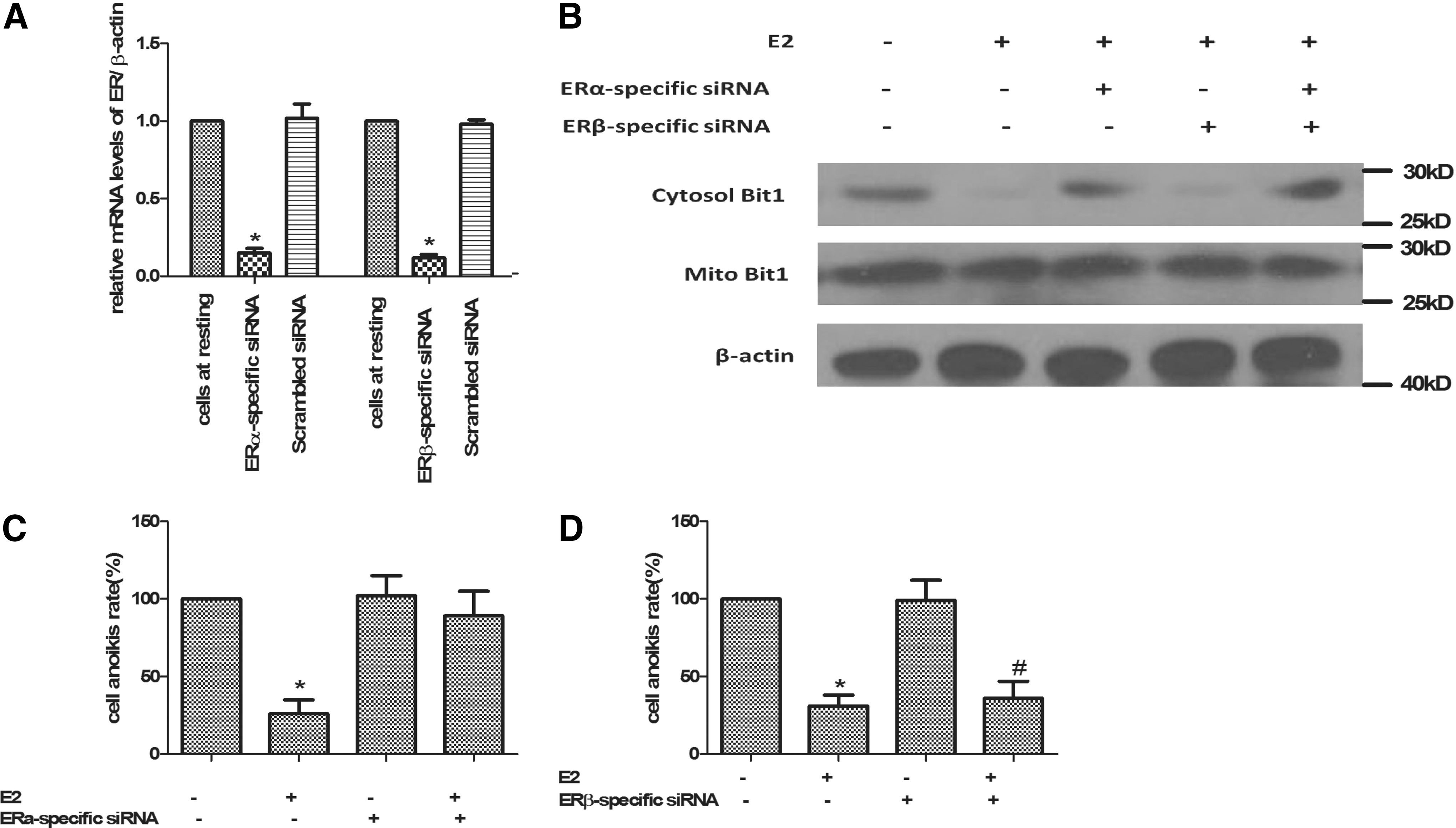
ERα, but not ERβ mediated the anoikis decreasing function of E2.
PI3K/AKT involvement in E2-induced decreasing of Caov-3 cell anoikis
It was reported that ERs binds to the p85α subunit of PI3K in breast cancer cells and vascular endothelial cells (Simoncini et al., 2000; Sun et al., 2001). PI3K phosphorylates the D-3 position of the phosphatidylinositol ring, catalyzing the synthesis of lipid mediators that serve as second messengers transferring the signaling cascade to intracellular protein kinases. One of the principal targets of this cascade is Akt. The activation of Akt mediates many of the downstream cellular effects of PI3K, including activation of cell survival pathways (Franke et al., 1997). Estrogen is known to stimulate the activity of Akt, which plays a central role in promoting the survival of a wide range of cell types (Zhang et al., 2001). There was evidence that the E2-induced proliferation in several cell lines requires ERα and the rapid phosphorylation of ERK and AKT (Marino et al., 2003); whereas E2, in the presence of ERβ, drives the cells out of the cell cycle by the proapoptotic p38/MAPK pathway activation (Acconcia et al., 2005). Thus, we evaluated these signaling functions in E2-induced anoikis and decreasing of cytosol Bit1 release. PI3K/AKT inhibitors or p38/MAPK inhibitors were added to cell cultures 0.5 h before E2 treatment; then cell anoikis and Bit1 level in cytosol were detected. The cytosol Bit 1 level was significantly increased by the addition of selective Akt inhibitor (CCT128930) compared with E2-treated-only group, but still lower than cells at resting. Whereas the addition of selective p38 inhibitor (CY2228820) only had no similar results (Fig. 4A). Figure 4B demonstrated that CCT128930 or CY2228820 treated only could slightly increase Caov-3 cell anoikis, whereas in cells cotreated with CCT128930 and E2, the reduction in cell anoikis due to E2 was reversed. CY2228820 had no similar effect on E2-induced cell anoikis reduction. All these suggested that PI3K/AKT were involved in the E2-induced decrease of Caov-3 cell anoikis.
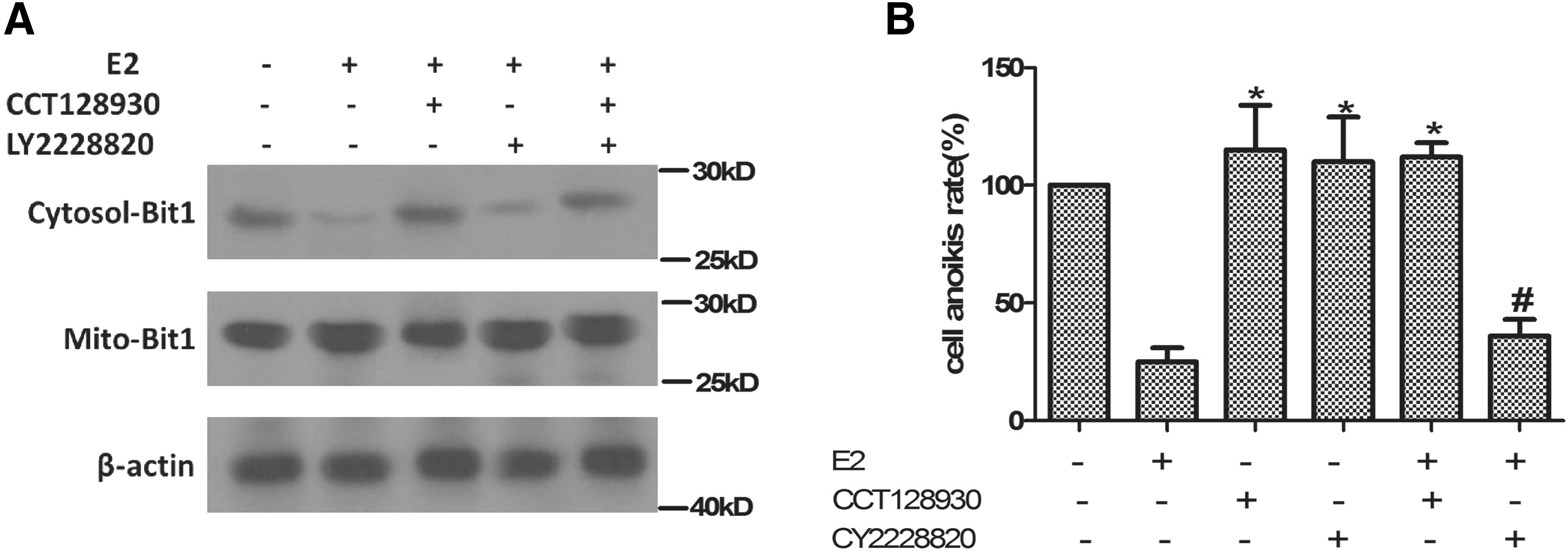
PI3K/AKT were involved in the E2 induced decrease of Caov-3 cell anoikis.
Discussion
In this study, we have described that treatment with E2 can reduce Caov-3 anoikis. Anoikis is defined as apoptosis that is induced by inadequate or inappropriate cell–matrix interactions (Frisch and Screaton, 2001). It is involved in a wide diversity of tissue homeostatic, developmental, and oncogenic processes such as cancer. Tumor cells are more resistant to anoikis and this contributes to tumor metastasis and invasion. So research on reducing tumor cell anoikis would result in important innovative therapies for the treatment of cancer. In the present study, we used suspended Caov-3 cells undergoing apoptosis mimicked anoikis process. Whereas treatment with E2, anoikis of Caov-3 reduced. This suggested that estrogen treatment may have promotion effects on ovarian tumor metastasis and invasion. Ovarian cancer causes more deaths from cancer than any other type of gynecological cancers. Most ovarian cancer patients do not respond to chemotherapy with complete remission or relapse after such treatment, so the use of estrogen therapies still remains an option for the treatment of ovarian cancer.
The regulation of apoptosis, including anoikis is under the influence of a wealthy set of genes. Most of them are proapoptotic and very few antiapoptotic. Among them, the bcl-2 family is of great importance since it contains proapoptotic and antiapoptotic members. Bit1, as Bcl2-inhibitor of transcription1, plays a critical role in apoptosis. Previous studies have shown that the release of Bit1 from mitochondria to cytosol can elicit anoikis (Jan et al., 2004; Griffiths et al., 2011), and downregulation of Bit1 can enhance metastasis of tumor cells (Karmali et al., 2011). These previous results and our findings raised the possibility that Bit1 was the executor of decreasing anoikis of Caov-3 cells treated with E2. Our results showed that cytosol level of Bit1 in E2-treated cells decreased whereas the mitochondrial level of that increased and the total level had no change. These suggested that E2 could inhibit the release of Bit1, which in turn reduced anoikis. Reduced anoikis of ovarian cancer cells may relieve metastasis of tumor cells, which was an indispensable step in tumor development. Thus our findings proposed that estrogen therapy may have a negative effect on ovarian cancer development, in a way, by reducing tumor cell apoptosis. This further fuels the concern of estrogen therapy on ovarian cancer.
To well understand the E2 mechanism, we also analyzed the downstream molecular pathway of E2. Studies have shown that the effect of E2 was mainly mediated by ERα and ERβ, but the two receptors' distributions are various from tissues and cell types. We used siRNA interference to detect the associated ER. Results revealed that E2 treatment exclusively through an ERα-dependent pathway led to decreasing of ovarian cancer cell anoikis. Studies from Halon et al. (2011) revealed that high level of ERα correlated with longer survival times, thus ERα expression may be a predictor of prognosis/survival in ovarian cancer patients. These findings all suggested the ERα may influence ovarian cancer through several ways, and the precise role of ERα is still open for research.
In this study, we further investigated the signaling pathways that may be involved. As is known to us, two of the main signaling pathways involved in cell survival are the Akt and NFκB signaling pathways. The PI3K/Akt/protein kinase B (PKB) pathway plays a central role in a variety of cellular processes, including cell growth, proliferation, motility, and survival in both normal and tumor cells. And there is also evidence that ERα is associated with PI3K/Akt (Marino et al., 2003), and E2 treatment significantly reduces AKT phosphorylation in MCF-7 beclin-overexpressing cells (John et al., 2008), so we generated the hypothesis that PI3K/Akt were the downstream molecules of ERα, and our results proved that. When PI3K/Akt inhibitors were added to cell cultures, E2's effect on anoikis was ameliorated, but not completely vanished. This suggested that PI3K/Akt pathway was not the only one that mediated the function of ERα. Further investigation exploiting lessons learnt from the whole signaling pathways will have a direct impact on the estrogen therapy of ovarian cancer.
In summary, the discovery of a new biology of E2-induced anoikis decreasing provides a drawback on estrogen in the treatment of ovarian cancer. Indeed, if a study of the molecular biology of estrogen-induced anoikis decreasing can define the mechanism precisely, then the molecules and signal pathways involved will become the promising target for a new group of ovarian cancer.
Footnotes
Disclosure Statement
The authors have no financial conflicts of interest.