
Abstract
HAX-1, a multifunctional protein involved in the regulation of apoptosis, cell migration, and calcium homeostasis, binds the 3′ untranslated region motifs of specific transcripts. This suggests that HAX-1 plays a role in post-transcriptional regulation, at the level of mRNA stability/transport or translation. In this study, we analyze in detail HAX-1 colocalization with processing bodies (P-bodies) and its dependence on mRNA availability. Endogenous P-body markers DCP1 and Rck/p54 were shown to colocalize with endogenous HAX-1, but in case of the overexpressed proteins, only DCP1 displayed unperturbed colocalization with HAX-1. HAX-1 colocalization with DCP1 was observed in most of the cell lines studied, but its presence was not required for P-body formation, and its silencing caused an increase in P-body number. Preliminary mapping suggested that HAX-1 has more than one short P-body-targeting sequence. The pools of P-body-localized HAX-1 and cytosolic HAX-1 were demonstrated to dynamically exchange, suggesting steady flow of the protein. Active transcription was shown to be a factor in the localization of HAX-1 to P-bodies. Also, it was observed that HAX-1 localizes to some unidentified foci, which do not contain DCP1. In addition, it was demonstrated that HAX-1 status influences vimentin expression levels. Overall, HAX-1 was shown to colocalize with P-body markers and influence P-body number per cell in a manner dependent on mRNA availability. Presented data support the hypothesis that HAX-1 is involved in mRNA processing as an element of P-body interaction network.
Introduction
HAX-1
In the current study, we analyzed the presence of HAX-1 in processing bodies (P-bodies), which are dynamic aggregates of translationally silenced mRNAs and proteins involved in mRNA degradation, translational repression, and other processes implicated in post-transcriptional regulation of gene expression. HAX-1 colocalization with P-bodies was briefly mentioned in our previous report (Grzybowska et al., 2013); in this study this colocalization is analyzed in detail. HAX-1 colocalized with P-body markers (DCP1 and Rck/p54), which supports the hypothesis that HAX-1 is involved in mRNA regulation. It was shown to be dispensable for P-body formation; moreover, its silencing caused a significant increase in P-body number per cell. The pools of P-body-localized and cytoplasmic HAX-1 were interchangeable. HAX-1 did not influence P-body motility, despite its actin-binding properties. Active transcription was required for HAX-1 to localize in P-bodies. Inhibition of active translation affected the localization of HAX-1 in P-bodies variably, depending on the inhibitor used, but generally tended to perturb the localization of HAX-1 in P-bodies. In addition, it was shown that HAX-1 silencing causes alternations of the vimentin expression levels. Taken together, these data indicate that HAX-1 is a P-body protein, but not essential for P-body formation and that its localization in P-bodies depends on intracellular mRNA levels. These results shed a new light on the cellular functions of HAX-1 and suggest it is involved in post-transcriptional regulation of gene expression.
Materials and Methods
Plasmids and constructs
Constructs containing the four human HAX-1 variants fused to GFP, and the constructs containing HAX-1-HA (hemagglutinin) were generated as described in Grzybowska et al. (2013). HAX-1 deletion constructs were generated by PCR amplification of the entire pEGFP-N1-HAX-1 (variant I) plasmid sequence apart from the regions that were to be deleted. Primers 5′-GAGAGTGATGCAAGAAGTGAATC-3′ and 5′-GCCGAAGTTATCGTGGAAACG-3′ or 5′-AGCAGTCCTAGGGGTGATCC-3′ and 5′-TTTGGGCTGGGGCTGTAGAAC-3′ were used to generate deletions Δ81–146 and Δ206–249, respectively. PCR was performed using the PfuPlus! high-fidelity DNA polymerase (EURx) and the following conditions: 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 6 min, and finally, 72°C for 7 min. The products were blunt-end ligated. The four proline residues (P253A, P256A, P258A, and P259A) were mutagenized using the primers 5′-GTCCTAGGGGTGATgCAGAATCAgCAAGAgCTgCAGCCCTG-3′ (forward) and 5′-CAGGGCTGcAGcTCTTGcTGATTCTGcATCACCCCTAGGAC-3′ (reverse) (mutated nucleotides are indicated in lower case letters), using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions.
The expression constructs for primary neuron transfection, pSyn-GFP and pSyn-Cherry, were obtained by amplifying and cloning synapsin-1 promoter into vectors pEGFP-N1 (Clontech) and pmCherry (Clontech), replacing the CMV promoter. Primers for promoter amplification were used as described in Dziembowska et al. (2012). To generate pSynHAX-1-GFP, HAX-1 was cloned from pEGFP-N1-HAX-1-GFP into pSynGFP using the HindIII and BamHI restriction sites. To generate pSynDCP1-Cherry, DCP1 was amplified from the mRFP-DCP1a template (described below) using the primers 5′-GAATTCAAGATGGCGCTGAGTCG-3′ (forward) and 5′-CTCGAGTGCTCCAGTCCTAGGTTGTG-3′ (reverse) and then cloned into pSynCherry between the EcoRI and XhoI restrictions sites.
To obtain FLAG-tagged human DCP1, coding sequence of DCP1 was amplified by PCR with AccuPrime Pfx DNA Polymerase (Invitrogen) and the following forward 5′-GAATTCATGGAGGCGCTGAGTCGAG-3′ and reverse 5′-CTCGAGTCATAGGTTGTGGTTGTCTTTG-3′ primers. The PCR product was A-tailed using Taq DNA Polymerase (Invitrogen), cloned into the pGEM-T Easy vector (Promega), and subcloned into the EcoRI site of pCR3-FL2 bearing two consecutive copies of the FLAG epitope preceding DCP1a ORF.
Constructs containing P-body markers were obtained from Paul Anderson, Harvard Medical School (mRFP-DCP1a) and from Dominique Weil, Andre Lwoff Institute, Paris, France (pCMV-SPORT6-rck/p54, RFP-tagged).
Cell culture and transfection
The HeLa (cervical carcinoma), MCF7 (breast cancer), MDA-MB-231 (breast cancer), MDA-MB-435 (melanoma), and Jurkat (T-cell leukemia) human cell lines were used, as well as rat primary neurons. Cells were grown in Dulbecco's modified Eagle's medium (HeLa, MCF7, MDA-MB-231, and MDA-MB-435) or RPMI (Jurkat) supplemented with 10% fetal bovine serum (Invitrogen). Cells were transfected using Lipofectamine (Invitrogen) according to the manufacturer's instructions. Jurkat cells were plated at a density of 300,000 cells/cm2 on coverslips coated with poly L-lysine (Sigma). Primary rat hippocampal neurons from P0 rat pups were dissociated using trypsin and plated on poly L-lysine-coated coverslips at a density of 50,000 cells/cm2 in Neurobasal A supplemented with B27, 2 mM glutamine, and 10 IU/mL penicillin–streptomycin. Hippocampal neurons at 10 days in vitro were transfected with pSyn-DCP1-RFP and pSyn-HAX-1-GFP using Lipofectamine (Invitrogen).
Fluorescence microscopy, live cell imaging, and data analysis
Standard immunofluorescence
Cells were grown on Lab-Tek standard coverslips (Nunc) and transfected with different constructs as indicated. Twenty-four hours after transfection (or 48 h in the case of primary neurons), cells were fixed in 4% formaldehyde, washed with the PEM buffer (80 mM PIPES, 5 mM EGTA, and 2 mM MgCl2), quenched with 0.1 M ammonium chloride, and permeabilized with PEM buffer containing 0.5% Triton X-100. To detect endogenous proteins, immunostaining was performed using mouse antibodies for HAX-1 (1:100; BD Biosciences), and RCK (1:50, E-8; Santa Cruz,) with goat anti-mouse IgG-FITC (1:200; Sigma) as a secondary antibody. Endogenous HAX-1 was also detected with the antibody produced in rabbit. The rat Hax-1 protein was overexpressed in E. coli and purified on Ni-NTA resin as in Sarnowska et al. (2007). The purified protein was sent to a commercially available source to produce an antibody. Obtained serum from rabbit was tested on Western blot to verify its specificity and subsequently purified using the ImmunoPure IgG Purification Kit (Pierce) according to the manufacturer's instructions.
Endogenous DCP1 was detected using the rabbit anti-DCP1A antibody (1: 100; Abcam) and anti-rabbit IgG-FITC (1:200; Life Technologies). Samples were blocked with TBS containing 5% low-fat milk and 0.1% Tween-20 for 30 min, incubated with a primary antibody at 4°C overnight, washed five times with a blocking buffer, incubated with a secondary antibody for 30 min, washed five times with TBS containing 0.1% Tween-20, and washed once with the PEM buffer. As a control, cells were labeled with the secondary antibody only. Cells were stained with DAPI, mounted, and observed on a Nikon Eclipse E-800 microscope with a 100× oil immersion objective (Plan Fluor; Nikon) using standard settings. The Intensity Correlation Analysis plugin of ImageJ image processing software (Abramoff et al., 2004) was used to quantify colocalization. The P-body-containing rectangle of the fixed area was selected and cropped as a separate image. Images were split into separate color channels, and the green and red channels were analyzed. The procedure was repeated for ∼50–100 P-bodies from 10 to 30 cells. The mean Pearson's colocalization coefficient (PCC) was calculated for each experiment. Statistical analysis of the PCC values was performed using the Student's t-test, following by the Fisher transformation.
Alternatively, cells were observed using a Leica TCS SP2 confocal laser scanning inverted microscope with a Plan Apo 63×1.4 NA oil immersion objective. Images were acquired at 0.5 μm Z-steps and the maximum projection of the images is presented.
Live cell imaging
HeLa cells were grown on Lab-Tek chambered coverglasses (Nunc) and transfected. Cells were observed using a Leica TCS SP5 confocal laser scanning inverted microscope and a Plan Apo 63×1.4 NA oil immersion objective in a chamber at 37°C containing 5% CO2. The GFP was excited at a wavelength of 488 nm and emitted light was detected at 500–550 nm. The RFP was excited at a wavelength of 561 nm using a DPSS diode laser and emitted light was detected at 570–620 nm.
Fluorescence recovery after photobleaching (FRAP) was performed on medium-sized P-bodies that were visually separate and in which GFP-HAX-1 and DCP1-RFP colocalized. Five baseline images of the cell were collected, and then the region of interest was scanned five times using high intensity 488 nm illumination sufficient to photobleach >90% GFP fluorescence. Typically, 40 images were then recorded at 2-s intervals using normal laser intensity. A 100 mW multiline argon laser (488 nm) at outputs of 5% and 80% was used for imaging and photobleaching, respectively. Single confocal sections of cells before, during, and after photobleaching were analyzed using Leica Application Suite Advanced Fluorescence software and ImageJ image processing software. P-bodies were manually tracked to monitor the recovery of fluorescence. The relative fluorescence intensity was determined, as previously described (Phair and Misteli, 2000), according to the following equation: relative fluorescence intensity=T 0 I f/T t I 0, where T 0 is the total fluorescence intensity of the cell before photobleaching, T t is the total fluorescence intensity of the cell at time t, I 0 is the fluorescence intensity of the bleached area before photobleaching, and I f is the fluorescence intensity of the bleached area at time t. Exponential curve fitting to determine the recovery half-time and plateau level was performed using Mathematica 5.2 (Wolfram Research).
To track P-bodies, cells were imaged in three dimensions (5 stacks, 60 frames, 140 s total time) and analyzed at maximum projection. Time-course analysis of P-body movement was performed using the Manual Tracking plugin of ImageJ. For each P-body, the mean square displacement (MSD) was calculated from XY pixel positions in all 60 frames (Shav-Tal et al., 2004; Aizer et al., 2008) and converted to μm using a Microsoft Excel macro created specifically for this purpose. P-body mobility distributions as assessed from the MSDs were compared using the Kolmogorov–Smirnov test (Statistica 6.0; StatSoft, Inc., OK).
Silencing of HAX-1 and Western blots
HAX-1 was silenced in HeLa cells using the BLOCK-iT Pol II miR RNAi Expression Vector system (Invitrogen). The miRNA sequence (5′-CCAAATCCTATTTCAAGAGCA-3′) was chosen to target HAX-1 variants I–IV. A double-stranded oligonucleotide encoding pre-miRNA was prepared and cloned into the pcDNA6.2-GW/EmGFP-miR vector according to the manufacturer's instructions. HeLa cells were transiently transfected with the silencing plasmid using Lipofectamine 2000 (Invitrogen). pcDNA6.2-GW/EmGFP-miR-neg plasmid (Invitrogen) was used as the negative control.
The HAX-1-silenced HeLa cell line was generated as described in Grzybowska et al. (2013). The level of HAX-1 protein was assessed by Western blotting. Total protein extract (65 μg) was separated on a 12% SDS-PAGE gel and transferred onto a Hybond-C Extra membrane (Amersham Biosciences). Membranes were blocked in PBS containing 5% low-fat milk and 0.1% Tween-20, incubated with a mouse monoclonal anti-HAX-1 primary antibody (1:250; BD Biosciences), incubated with an HRP-conjugated goat anti-mouse secondary antibody (1:10,000; Pierce), and developed using enhanced chemiluminescence (Amersham Biosciences) according to the manufacturer's instruction. To confirm equal protein loading, blots were reprobed with a rabbit polyclonal anti-GAPDH primary antibody (1:10,000; Sigma) and a polyclonal HRP-conjugated goat anti-rabbit secondary antibody (1:10,000; Abcam). Western blots for vimentin were performed, as above, using the mouse monoclonal anti-vimentin antibody (1:200; Sigma).
qPCR
Total RNA and cDNA from HeLa cells (0.5×106 cells per repeat) were obtained using the PureLink RNA Mini Kit (Life Technologies) and SuperScript III (Life Technologies), respectively. qPCR assays for vimentin mRNA levels were performed using SYBR Green PCR Master Mix (Applied Biosystems) with primers forward: 5′-ACAACCTGGCCGAGGACATC-3′ and reverse: 5′-CCTCTTCGTGGAGTTTCTTC-3′ amplifying 194 bp and, as a reference, 226 bp of the GAPDH transcript (forward: 5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse: 5′-GAAGATGGTGATGGGATTTC-3′). PCR was performed in the following conditions: precycling hold at 95°C for 10 min, cycles: 95°C, 30 s, 60°C, 60 s, up to 40. The ΔΔCT method was used for quantity calculations.
Coimmunoprecipitation
Constructs encoding DCP1-FLAG and HAX-1-HA were cotransfected into HeLa cells using Lipofectamine 2000 (Invitrogen). HeLa cells cotransfected with the empty pCR3-FL2 vector and HAX-1-HA were used as controls. Cells were lysed with the RIPA buffer supplemented with protease inhibitors (PI; Roche) and cleared lysates subsequently incubated with EZview Red ANTI-FLAG M2 Affinity Gel (Sigma) overnight at 4°C. Beads were extensively washed with RIPA+PI (four times, 5 min, 4°C) and the immunoprecipitated proteins extracted by incubation with the Laemmli loading buffer at 98°C, 10 min. The immune complex was separated by electrophoresis on a 10% SDS-polyacrylamide gel and electrotransferred to a modified nitrocellulose membrane (Amersham) for Western blot analysis (West Pico; Pierce). The antibodies used were as follows: anti-HA primary antibody (rat, 1:250; Sigma), peroxidase-conjugated secondary antibodies (anti-rat, 1:5,000; Sigma), ANTI-FLAG M2 mouse monoclonal antibody, HRP conjugated (1: 1000; Sigma).
Results
HAX-1 colocalizes with DCP1 and Rck/p54 in HeLa cells and alters P-body dynamics
In agreement with our previous report (Grzybowska et al., 2013), the cytoplasmic granular structures in HeLa cells in which HAX-1-GFP concentrated were identified as P-bodies by colocalization with the P-body marker, DCP1. This colocalization was confirmed for both, endogenous and overexpressed proteins (Fig. 1A). Colocalization was also examined for HAX-1 and the RNA helicase Rck/p54 (also known as DDX6), which is another essential P-body protein. Whereas endogenous HAX-1 and Rck/p54 colocalized, concomitant overexpression of HAX-1-GFP and Rck/p54-RFP resulted in almost no colocalization (Fig. 1B).
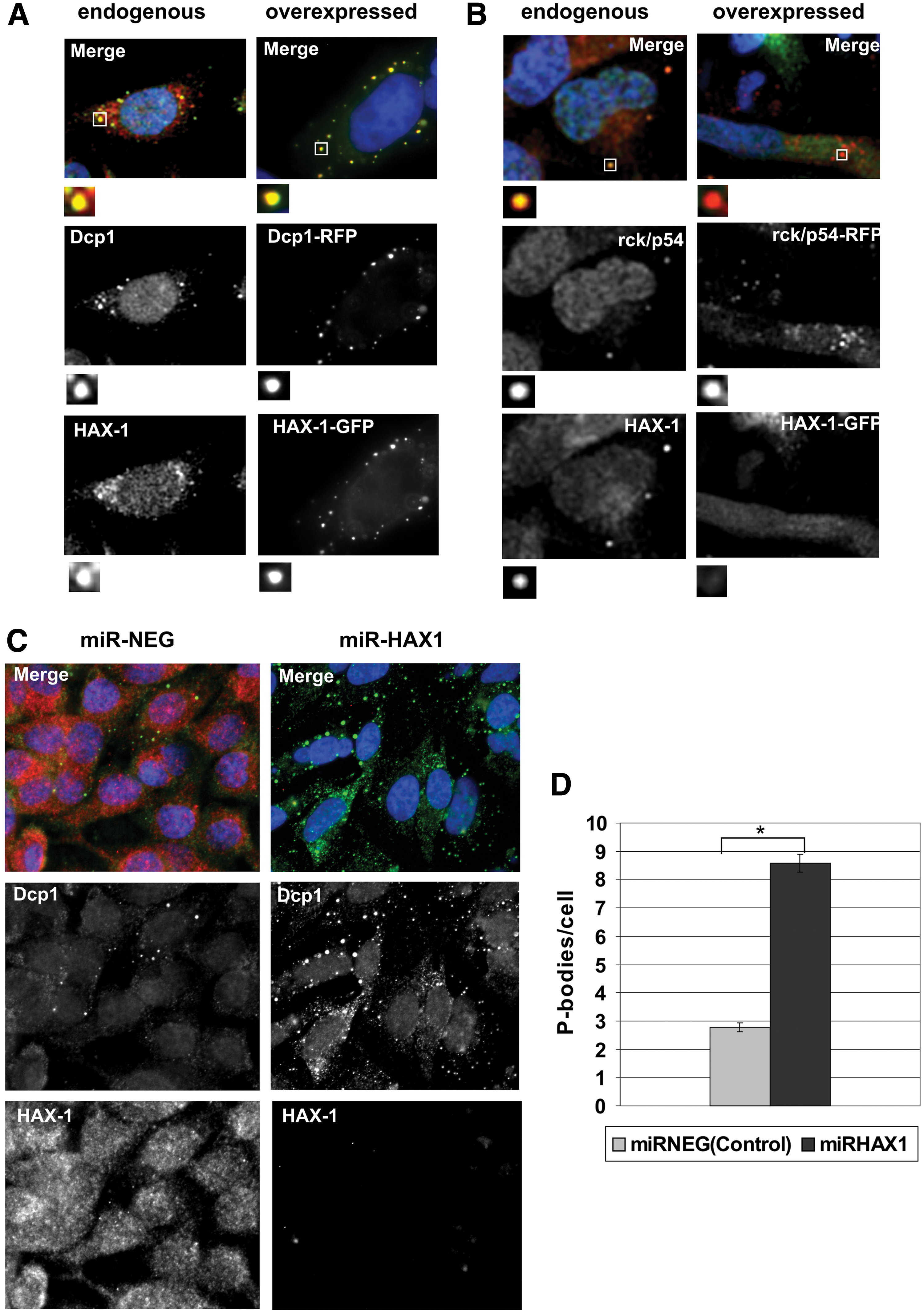
HAX-1 colocalizes with processing body (P-body) markers and influences P-body number.
As reported previously (Grzybowska et al., 2013), P-bodies were still present following HAX-1 knockdown, demonstrating that HAX-1 is not required for P-body formation. Furthermore, the effect of HAX-1 silencing on P-bodies was quantified, revealing a significant increase in P-body number per cell in a stable HAX-1-silenced cell line (Fig. 1C, D). Silencing of HAX-1 was verified by Western blotting (Supplementary Fig. S1; Supplementary Data are available online at
HAX-1-DCP1 colocalization is hard to disrupt
The HAX-1 gene is alternatively spliced, producing eight splice variants in human cells. Variants V–VIII are expressed at very low levels and contain frameshifts that change the protein sequence (Lees et al., 2008; Trebinska et al., 2010); therefore, only variants I–IV were analyzed in this study (Fig. 2A, B). DCP1-RFP colocalized with variants I–III; however, it colocalized significantly less with variant IV. DCP1-RFP colocalized slightly less with variant II than with variant I; however, the difference was borderline significant. The degrees of colocalization between DCP1-RFP and the various GFP-tagged HAX-1 variants were measured by using the ImageJ colocalization algorithm and are presented as PCCs in Figure 2C. Moreover, colocalization with DCP1-RFP was also analyzed for three GFP-tagged mutant HAX-1 proteins: two large deletions, encompassing regions in N- and C-terminal parts of the protein and the mutation of the proline motif (P253A, P256A, P258A, and P259A) present at the C-terminal end of the HAX-1 sequence (Supplementary Fig. S2A). None of these mutants exhibited significantly reduced colocalization with DCP1-RFP. Decrease of colocalization for the proline mutant (7) was borderline significant as well as its increase for the C-terminal deletion mutant (6) (Fig. 2B, C).
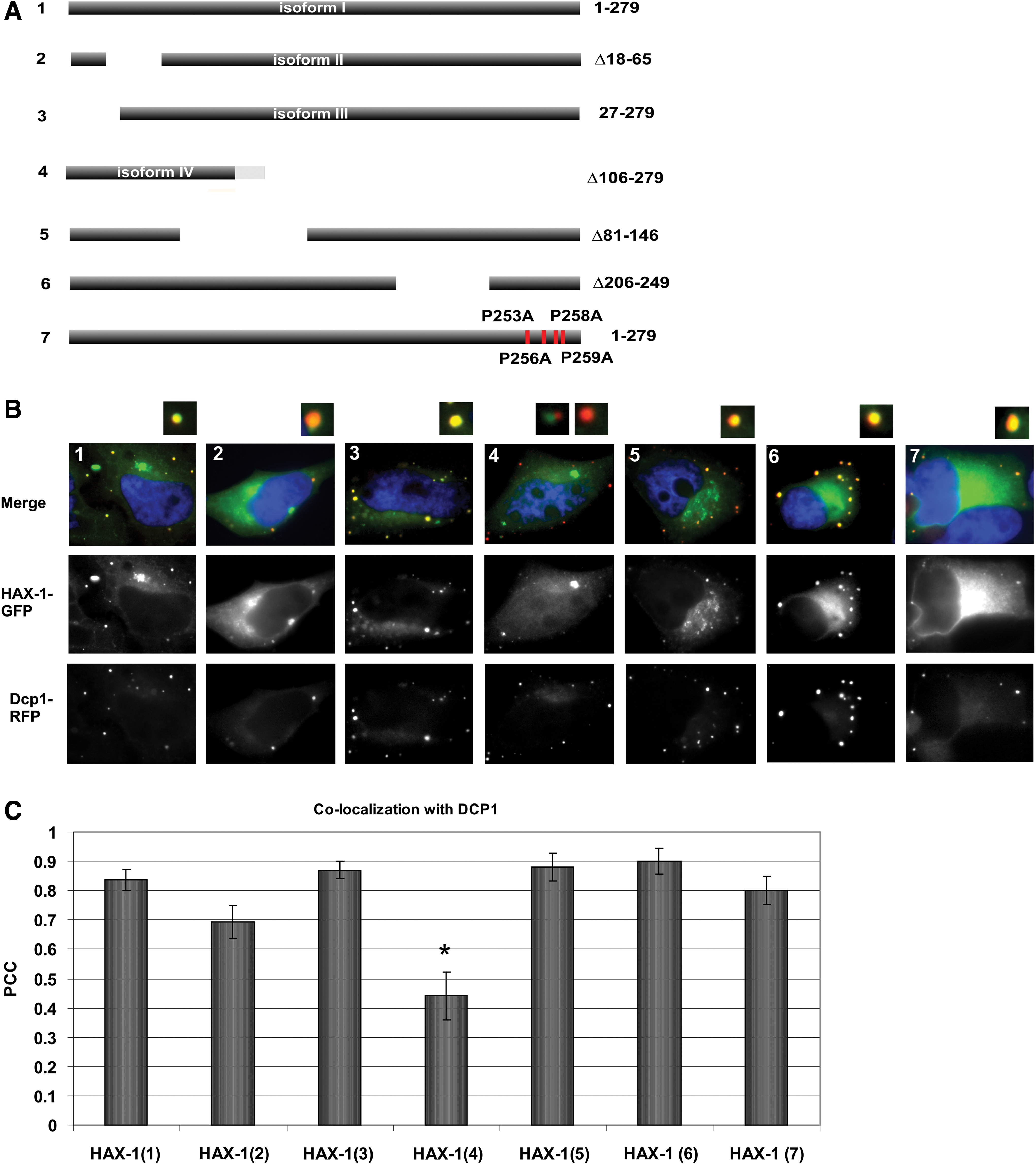
Colocalization between various GFP-tagged HAX-1 constructs and DCP1-RFP.
Proline-rich motif followed by the predicted alpha-helical structure on the C-terminus of HAX-1 resembles proline-rich motif in Drosophila XRN1 nuclease, which is responsible for DCP1 binding (Braun et al., 2012). A comparison of the two motifs is presented in Supplementary Figure S2A. In addition, to test the possibility of the direct interaction between HAX-1 and DCP1, coimmunoprecipitation with tagged proteins HAX-1-HA and DCP1-FLAG was performed, but the result was negative (Supplementary Fig. S2B). Also, mutation of the four proline residues (PXXPXPP) of HAX-1 motif had only a minimal effect on its colocalization with DCP1-RFP (Fig. 2B7, C7).
Colocalization between HAX-1 and DCP1 is present in different cell types
To verify if HAX-1-DCP1 colocalization is tissue specific, colocalization between HAX-1-GFP and DCP1-RFP was examined in several different cell lines (Fig. 3), namely, two breast cancer cell lines with different invasive potentials (the epithelial MCF-7 cell line and the mesenchymal MDA-MB-231 cell line), a melanoma cell line (MDA-MB-435), T-lymphoblastoid cell line (Jurkat), and primary neurons. HAX-1-GFP and DCP1-RFP colocalized in all these cell lines, apart from primary neurons in which HAX-1-GFP localized to foci that were not labeled with DCP1-RFP (Fig. 3).

Colocalization between HAX-1-GFP and DCP1-RFP in various cell types. Cells were fixed 24 h after transfection with HAX-1-GFP and DCP1-RFP. Color images available online at
HAX-1 as a P-body component: influence on P-body movement and the dynamics of its localization
Since HAX-1 interacts with the actin cytoskeleton and binding to actin influences the movement of P-bodies, time-course analysis was performed to determine whether knockdown of HAX-1 affects the movement of P-bodies (Fig. 4A and Supplementary Video S1). About 100 P-bodies were tracked (60 frames, 140 s) in each experiment and the MSD of each P-body was calculated. The Kolmogorov–Smirnov test showed that the cumulative distributions of P-body movement in control cells (transfected with pcDNA6.2-GW/EmGFP-miR-neg) and HAX-1-silenced cells were not significantly different (Fig. 4B).

HAX-1 does not influence P-body movement, and cytoplasmic and P-body-associated pools of HAX-1 are dynamically exchanged.
FRAP experiments were performed to establish whether HAX-1 resides permanently in P-bodies or whether it is dynamically exchanged between P-bodies and the cytoplasm. After photobleaching, HAX-1-GFP fluorescence was recovered in P-bodies with a recovery half-time of 24 s and the immobile fraction was about 45% (Fig. 4C, D and Supplementary Video S2).
Active transcription and translation are important for the effective HAX-1-DCP1 colocalization
HAX-1-GFP and DCP1-RFP colocalization was tested in the presence of several factors previously shown to influence P-body colocalization of other proteins or HAX-1 cellular localization. Glucose starvation was shown to alter P-body dynamics in Saccharomyces cerevisiae (Teixeira et al., 2005) and in HeLa cells (Takahashi et al., 2011). Sodium arsenite was applied because, as shown previously (Grzybowska et al., 2013), it causes nuclear translocation of HAX-1, which may be important for mRNA binding. However, none of these stress factors affected HAX-1 colocalization to P-bodies.
To assess the role of active transcription and translation for HAX-1-DCP1 colocalization, cells were treated with the transcription inhibitor actinomycin D and two translation inhibitors with different modes of action, puromycin and cycloheximide (CHX). HAX-1 and DCP1 colocalized significantly less in cells treated with actinomycin D than in untreated controls (Fig. 5A, B). In puromycin-treated cells, HAX-1-GFP partially colocalized with DCP1-RFP; however, this colocalization was perturbed in many cells and HAX-1 aggregated in cytoplasmic foci of unknown identity. These HAX-1-GFP-containing bodies often partially overlapped or were in close proximity to DCP1-RFP-containing P-bodies (Fig. 5A). The number of DCP1-RFP-positive foci that did not colocalize with HAX-1-GFP, the number of HAX-1-GFP-positive foci that did not colocalize with DCP1-RFP, and the number of foci in which DCP1-RFP and HAX-1-GFP colocalized were calculated in puromycin-treated and untreated cells. These data showed that the number of foci in which HAX-1-GFP and DCP1-RFP colocalized was markedly reduced following puromycin treatment (Fig. 5C). In CHX-treated cells, P-bodies dispersed and HAX-1-GFP and DCP1-RFP were diffusely distributed in most cells (Fig. 5A). In cells that still contained P-bodies following CHX treatment, DCP1-GFP and HAX-1-RFP mostly colocalized (not shown).
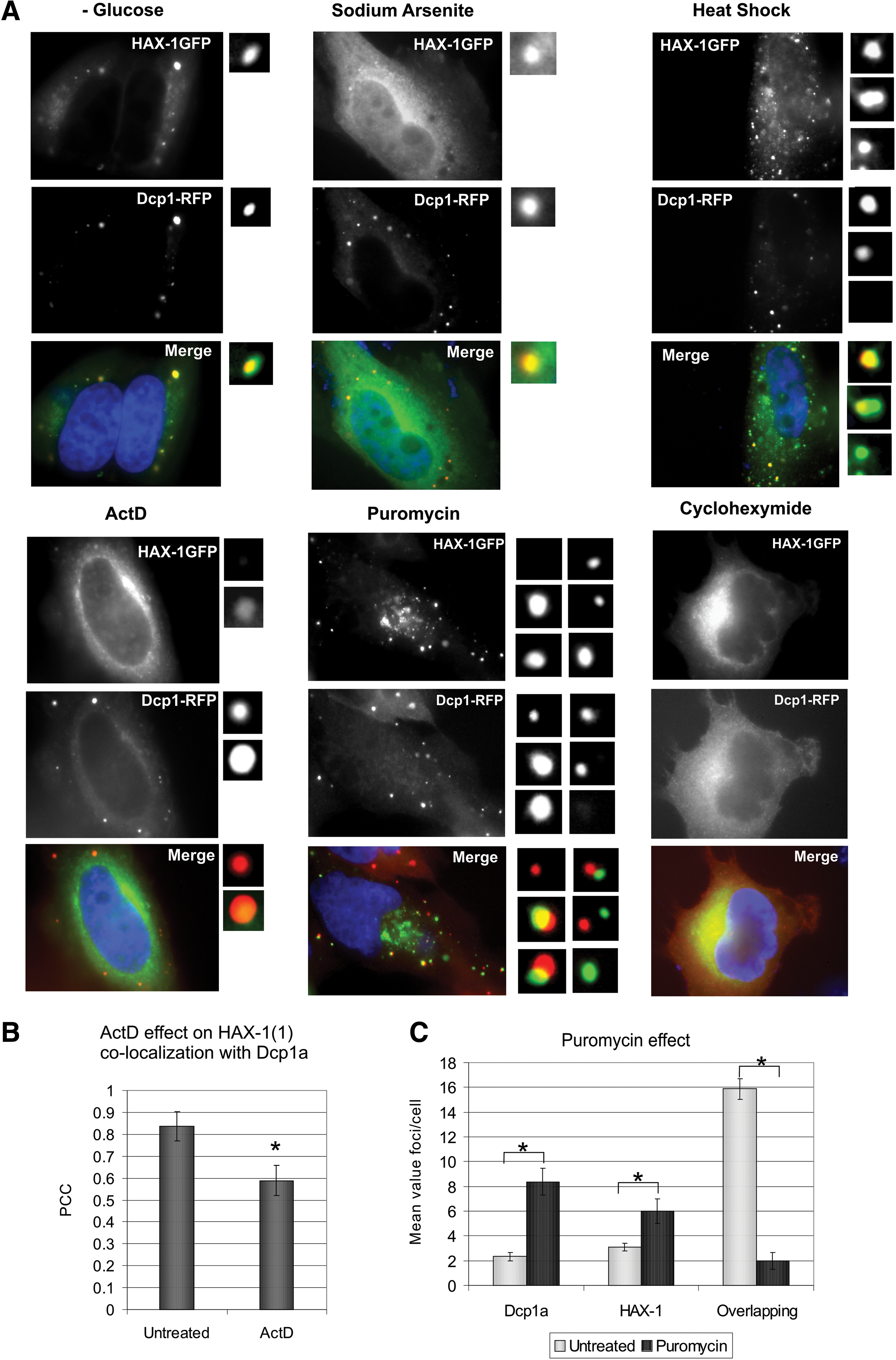
Active transcription and translation influence HAX-1 localization to P-bodies.
HAX-1 silencing alters vimentin expression levels
Vimentin mRNA represents one of the two transcripts that were shown to bind to HAX-1 (Al-Maghrebi et al., 2002), but HAX-1 influence on its expression was never investigated. In this study, we present the qPCR analysis of vimentin expression in HAX-1-silenced cells versus silencing control (Fig. 6A). HAX-1 silencing significantly reduced vimentin mRNA expression and this effect was even more pronounced in sodium arsenite-stressed cells. Sodium arsenite treatment was applied, because it influenced the expression levels of POLB transcript in the same cell lines (Grzybowska et al., 2013). In addition, vimentin expression was also tested in the studied cell lines at the protein level by Western blot analysis (Fig. 6B).
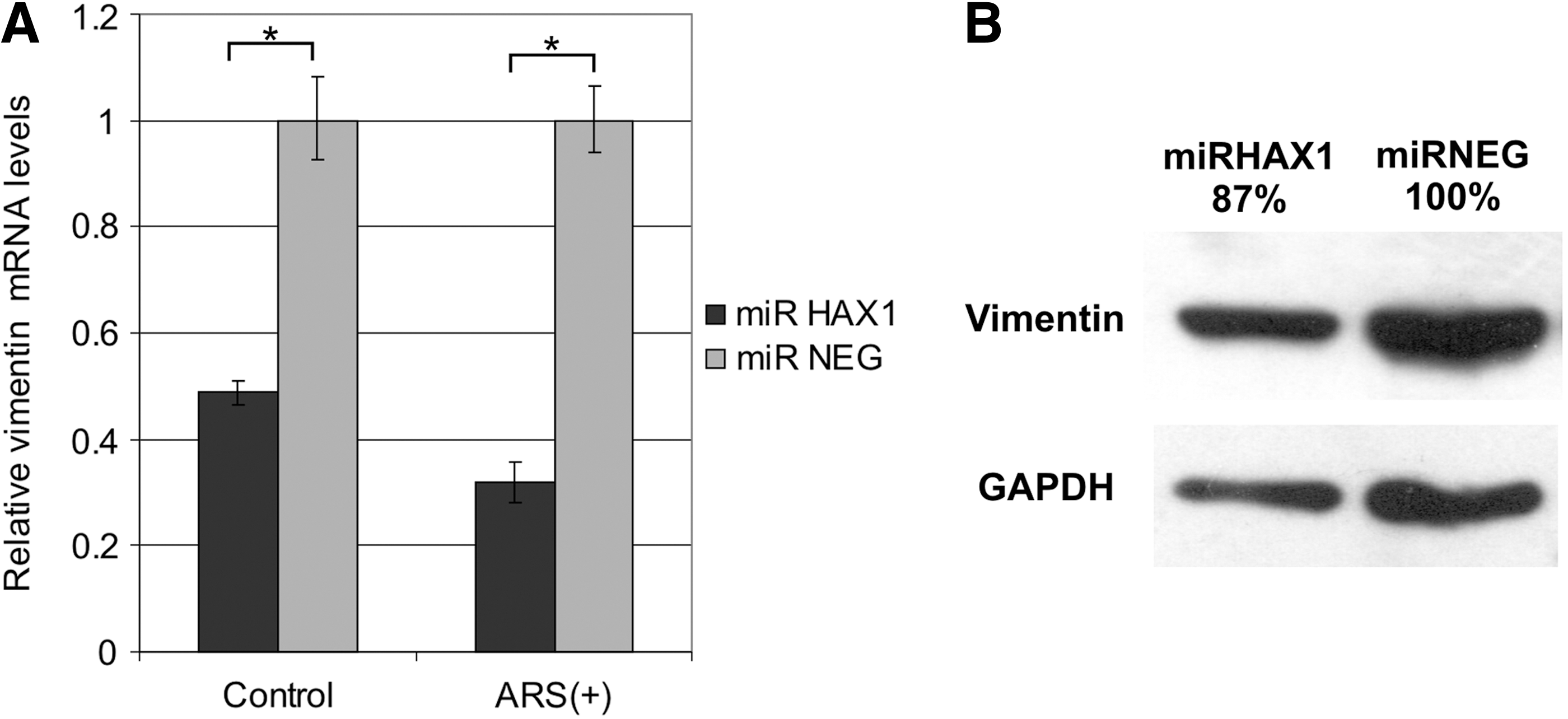
HAX-1 silencing affects vimentin expression levels.
Discussion
HAX-1 is implicated in crucial cellular processes such as apoptosis, migration, and the regulation of calcium content in the endoplasmic reticulum (Fadeel and Grzybowska, 2009). Despite these wide-ranging processes with which HAX-1 is associated, the mechanisms by which HAX-1 functions are unknown. The binding of HAX-1 to the 3′UTRs of specific transcripts and preliminary data of the effects of this on transcript stability (Grzybowska et al., 2013) indicate that HAX-1 is involved in the processing of mRNAs. This is supported by the localization of HAX-1 in P-bodies explored in detail in the current study. HAX-1 colocalized with DCP1, which is a decapping enhancer and a P-body marker. This colocalization was shown for HAX-1-GFP- and DCP1-RFP-overexpressing cells and for endogenous proteins. The DEAD box protein Rck/p54 is another P-body marker and important for the assembly of these foci. Rck/p54 colocalizes with DCP1 (Swetloff et al., 2009) and is a part of the decapping complex (Coller et al., 2001). Intriguingly, overexpressed HAX-1 colocalized strongly with overexpressed DCP1 but not with overexpressed Rck/p54. The apparent lack of colocalization of the overexpressed HAX-1 and Rck/p54 indicates that HAX-1 abundance may affect proper localization, possibly by the competition for the binding sites in the complex. It seems possible that DCP1 overexpression contributes to HAX-1 sequestering, because in HAX-1-overexpressing cells without concomitant DCP1 overexpression, colocalization is markedly less visible (data not shown). A plausible explanation could be that HAX-1 not only colocalizes with DCP1 but also is recruited through this protein into the decapping complex and, when overexpressed, competes with Rck/p54 for a binding site and impedes its incorporation into the complex. Direct interaction of HAX-1 with DCP1 seemed further probable because of the presence of the motif containing four proline residues (PXXPXPP). This motif is similar in terms of proline distribution and the presence of the predicted alpha-helical structure just behind this motif, to the DCP1-binding motif (DBM) of Drosophila XRN1 exonuclease (Braun et al., 2012), another component of the decapping complex. Human XRN1 is devoid of this motif and, since HAX-1 is present exclusively in Chordata, it could be hypothesized that it bridges DCP1 and XRN1 using its proline motif. However, mutation of this motif did not alter colocalization between HAX-1 and DCP1 and coimmunoprecipitation of the tagged proteins yielded negative results (Supplementary Fig. S2); hence, this hypothesis should be abandoned. However, these findings do not preclude the possibility that HAX-1 is a component of the decapping complex.
Several HAX-1 constructs were used to identify the sequence that targets HAX-1 to P-bodies. DCP1-RFP colocalized significantly less only with HAX-1 variant IV, which contains the first 106 amino acids of the protein. The colocalization between other HAX-1 deletion mutants and DCP1-RFP was only slightly affected, if at all. These results indicate that there are several short motifs responsible for HAX-1 targeting to P-body, located in different regions of the protein and, thus, hard to eliminate altogether.
Since HAX-1 interacts with the actin cytoskeleton (Gallagher et al., 2000) and P-bodies are immobile when bound to actin bundles (Aizer et al., 2008), we investigated the effects of HAX-1 overexpression and silencing on the motility of P-bodies. The P-body movement was unaffected by overexpression (data not shown) or silencing of HAX-1, suggesting that this protein is not responsible for P-body contacts with the actin cytoskeleton.
FRAP experiments established that HAX-1 is dynamically recruited into P-bodies from the cytoplasm. This, in combination with the localization of HAX-1 at various cellular locations and the finding that HAX-1 was not essential for P-body formation, indicates that HAX-1 is not a core P-body protein. The relatively long recovery half-time (24 s) and high immobile fraction (45%) indicate that HAX-1 is actively bound in P-bodies.
Colocalization between HAX-1-GFP and DCP1-RFP was significantly reduced following treatment with the transcription inhibitor actinomycin D. This suggests that targeting of HAX-1 to P-bodies requires active transcription, and so is linked to mRNA binding and processing. Two inhibitors of translation were used to establish if active translation has an impact on HAX-1 P-body localization: CHX, which blocks translation elongation and thereby arrests mRNA on ribosomes, and puromycin, which causes premature chain termination, releasing mRNA from ribosomes and generating an affluence of nontranslating mRNA in the cell (Cougot et al., 2004; Eulalio et al., 2007; Decker and Parker, 2012). CHX causes P-body dispersion; accordingly, HAX-1 was mostly diffusely distributed in the cytoplasm of CHX-treated cells. The effects of puromycin were different and intriguing. HAX-1-GFP and DCP1-RFP partially colocalized in puromycin-treated cells; however, many cells had foci that contained HAX-1, but not DCP1, and were similar to P-bodies in terms of size and appearance. These foci and P-bodies often partially overlapped and appeared to dock, possibly exchanging protein content. Foci containing HAX-1 but not DCP1 were observed in cells that were not treated with puromycin; however, puromycin markedly enhanced their number. The identity, protein content, and cellular roles of these foci remain unknown. It may be hypothesized that a high level of translationally incompetent mRNA induces the formation of HAX-1-containing bodies, and that these bodies have roles in the transport or storage of such mRNAs.
HAX-1 knockdown did not prevent P-body formation, indicating that this protein is not essential for this process, but on the contrary, it caused significant increase in P-body number per cell, suggesting that this protein affects P-body dynamics. A plausible explanation could be that HAX-1-containing bodies also contain some other P-body proteins (but not DCP1) sequestering them and affecting the overall number of DCP1 foci by competing for essential proteins. Further research is needed to prove or disprove this hypothesis.
Colocalization between HAX-1 and DCP1 was examined in several cell types, including primary neurons and lymphocytes. HAX-1-GFP and DCP1-RFP colocalized in each of the cell lines tested, apart from primary neurons. Intriguingly, as in HeLa cells treated with puromycin, two types of granules were observed in primary neurons: DCP1-containing P-bodies and granules containing HAX-1 but not DCP1. These results suggest different functions of HAX-1 in neurons, possibly linked to the regulation of mRNA transport and translation, since transport of translationally arrested mRNA in RNA granules and localized translation are common in these cells (Martin and Zukin, 2006). It remains to be established if there is a link between the HAX-1-containing granules observed in neurons and the granules observed in the puromycin-treated HeLa cells.
Supporting the notion of HAX-1 involvement in mRNA regulation, HAX-1 status was shown to influence vimentin mRNA levels. However, in contrast to previously demonstrated influence on POLB mRNA (Grzybowska et al., 2013), HAX-1 silencing caused a reduction in vimentin mRNA expression levels, further augmented by sodium arsenite treatment. This indicates that HAX-1 impact on mRNA is complex, and requires further studies.
Intriguingly, vimentin protein levels were also diminished by HAX-1 silencing, but not to the extent observed at the mRNA level. This indicates that HAX-1 may be involved in the intricate regulation encompassing mRNA stability and translational control.
Conclusions
Data presented in this study confirm that HAX-1 is a P-body protein and has a role in mRNA processing. Targeting of HAX-1 to P-bodies requires active transcription, and colocalization between HAX-1 and DCP1 is altered by the amount of nontranslatable mRNA. This suggests that HAX-1 is also involved in mRNA regulation, a conclusion supported by its influence on vimentin expression levels. Although the specific post-transcriptional processes in which HAX-1 functions are yet to be identified, HAX-1 P-body localization and its impact on specific mRNA levels indicates its involvement in the regulation of mRNA stability/transport and possibly translation.
Footnotes
Acknowledgments
The authors thank Antek Laczkowski for assistance in statistical analysis and Paul Anderson and Dominique Weil for the constructs. This work was supported by Polish National Science Centre (grants N301 317439, 2011/01/B/NZ1/03674, and 2012/07/N/NZ1/00104).
Disclosure Statement
The authors declare that they have no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.