
Abstract
This study aimed to determine the molecular mechanism by which the oncogenic micoRNA-21 (miR-21) functions in bladder cancer. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis showed that the expression of miR-21 considerably increased in primary cancer tissue compared with that in the paired adjacent noncancerous tissue and that in normal bladder mucosa. Knockdown of miR-21 by using antisense oligonucleotide significantly suppressed the proliferation and migration of bladder cancer cells (J82 and RT112). Mechanism studies showed that downregulation of miR-21 resulted in cell cycle arrest at the G1 phase and upregulation of the tumor suppressor PTEN (phosphatase and tensin homologue) and p53 phosphorylation at Ser46. The p53 phosphorylation at Ser15 and the whole level of p53 acetylation remained unchanged in response to miR-21 knockdown. MicroRNA-21 regulates proliferation and migration of bladder cancer cells and cross talk with PTEN and p53 in bladder cancer.
Introduction
W
MicroRNAs are small noncoding RNAs of ∼22 nucleotides. Binding of microRNA to the 3′ UTR of its target mRNA will consequently cause mRNA degradation or translational repression, thereby regulating gene expression at the post-transcriptional level (Ameres and Zamore, 2013). Numerous studies have demonstrated the involvement and regulatory roles of microRNAs in initiation, progression, and metastasis of tumors (Adams et al., 2014). Thus, microRNAs have been suggested as the targets for cancer therapy (Ling et al., 2013).
The microRNA-21 (miR-21) was initially found to be abnormally expressed in brain tumor (Chan et al., 2005). Substantial evidence has indicated that miR-21 plays an oncogenic role in many cancer types (Wang et al., 2014). Overexpression of miR-21 promoted cancer cell proliferation, migration, and metastasis (Xu et al., 2014), whereas downregulation of miR-21 exerted the opposite effects (Meng et al., 2007; Xiong et al., 2013). With regard to bladder cancer, microRNA array analysis revealed the remarkably raised ratio of miR-21/miR-205 in the invasive cell lines compared with the noninvasive cell lines (Neely et al., 2010). In another study, miR-21 was identified as the most elevated one among the upregulated microRNAs in bladder cancer tissue compared with normal bladder mucosa (Dyrskjot et al., 2009). Additionally, the association of miR-21 with high-grade tumor and poor prognosis of bladder cancer was also reported (Catto et al., 2009). Despite of our accumulating knowledge about miR-21, the oncogenic mechanism of miR-21 in bladder cancer remains elusive.
In the present study, we investigated the influences of knockdown of miR-21 in proliferation and migration of bladder cancer cells. We also analyzed the cross talk between miR-21 and the tumor suppressors, PTEN (phosphatase and tensin homologue) and p53, in bladder cancer cell lines.
Materials and Methods
Clinic samples
Bladder tumor tissue and the paired adjacent noncancerous tissue were obtained from patients who underwent radical cystectomy. Normal bladder mucosa samples were obtained from enucleation of the prostate of patients with prostatic hyperplasia. Written consents were received from patients for all clinic samples. The experiment procedures involving human samples were approved by the Medical Ethics Committee of Second Hospital of Tianjin Medical University.
Cell lines and culture conditions
The J82 cell line was obtained from American Type Culture Collection. The RT112 cell line was purchased from Sigma-Aldrich. J82 and RT112 cell lines were maintained in RPMI medium supplemented with 10% fetal bovine serum at 37°C and 5% CO2.
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA extraction with tissue or cells was conducted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. For analysis of miR-21 expression, the stem-loop reverse transcription primer, quantitative real-time polymerase chain reaction (RT-PCR) primers, and TaqMan MGB probe referenced to the sequences previously described (Chen et al., 2005; Song et al., 2010). The total RNA was used for cDNA synthesis with the Superscript II Kit (Qiagen). qRT-PCR was performed in compliance with a standard TaqMan PCR protocol using the ABI 7900HT System (Applied Biosystems). The relative level of miR-21 was determined by using the ΔCt method, and the expression level of U6 RNA served as the internal reference.
MTT assay
Cells in a six-well plate were transfected with 100 pmol per well of miR-21 antisense oligonucleotide (ASO) (5′-UCAACA UCAGUCUGAUAAGCUA-3′) or the control oligonucleotide (5′-CAUUAAUGUCGGACAACUCAAU-3′) by using Lipofectamine 2000 (Invitrogen). Twenty-four hours later, cells were transferred into a 96-well plate at 104 cells/well. After an incubation of another 48 h, 20 μL MTT (5 mg/mL; Sigma-Aldrich) was added to each well and the plate was incubated at 37°C with 5% CO2 for another 4 h. Then, the medium was removed by centrifugation and 100 μL dimethyl sulfoxide was added into each well. The absorbance at 490 nm of the solution was measured.
Flow cytometry analysis
Cells were harvested at 48 h after transfection with the miR-21 ASO or control ASO and washed twice with phosphate-buffered saline. For cell cycle analysis, cells were subjected to overnight fixation with 70% cold ethanol at 4°C, followed by RNase digestion and propidium iodide (PI) staining, before cells were analyzed with the BD LSRII Flow Cytometry System (BD Bioscience). For cell apoptosis assay, harvested cells were dual labeled by fluorophore-conjugated Annexin V and 7-AAD (SouthernBiotech).
Colony formation assay
Forty-eight hours after transfection with the miR-21 ASO or control ASO, cells were collected and seeded in a 12-well plate at 120 cells/well. Cells were cultured for another 10 days under standard conditions. To count the colony, cells were fixed with methanol for 10 min, followed by crystal violet staining for 10 min.
Western blot analysis
Cells were collected at 72 h upon transfection and lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) supplemented with proteinase and phosphatase inhibitor cocktails (Sigma-Aldrich). Protein samples (20 μg per lane) were loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) for separation, and then electrotransferred onto a nitrocellulose membrane (Millipore). The membranes were blocked in tris-buffered saline (TBS) buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl) with 5% milk and 0.02% Teween-20 at room temperature for 1 h. The primary antibody was incubated overnight at 4°C. HRP-conjugated secondary antibody (Jackson Immunoresearch laboratory) incubation was performed at room temperature for 1 h. The blots were developed with Western Lightning-Enhanced Chemiluminescence Substrate (Perkin Elmer) and exposed to X-ray films.
To detect the acetylation state of p53, 5×106 cells transfected with the miR-21 ASO or control ASO were lysed in 0.5 mL RIPA buffer, and then the supernatant was added with equal volume of TBS buffer before the pull-down of p53 by using the rabbit polyclonal antibodies. The immunoprecipitated p53 was probed with the mouse monoclonal antibody against acetylated lysine.
Specific antibodies against PTEN (cat#9559S), p53 (cat#2527S), phosphorylated p53 (ser15 cat#9286S and ser46 cat#2521P), and acetylated lysine (Ac-K-103, cat#9681) were from Cell Signaling Technology. The polyclonal antibodies against p53 (cat#sc-1311-R) for immunoprecipitation were from Santa Cruz Biotechnology. The primary antibody recognizing GAPDH was from Bioss.
Wound healing assay
Cells were seeded in a six-well plate to reach basically 100% confluence in monolayer the next day. The wound was made with a 1-mL micropipette tip, scraping the monolayer cells in a straight line. The detached cells were removed by washing twice with the medium. The closure of wound was observed under a phase-contrast microscope (Olympus).
Transwell assay
Ten milliliters of fibronectin (0.5 mg/mL; Sigma-Aldrich) were spread onto the bottom surface of the membrane in a transwell chamber (Corning) and left to air dry in a tissue culture hood. The chamber was coated with 50 μL Matrigel (BD Biosciences) and incubated for 40 min at 37°C. Forty-eight hours after transfection, cells were transferred to chambers at 105 cells/well and cultured in Opti-MEM medium (GIBCO). The lower chamber was filled with ECM medium (GIBCO). Sixteen hours later, nonmigrated cells were removed using cotton swabs. Migrated cells were fixed with 100% methanol for 30 min and stained by crystal violet for 15 min. Cell images were captured under a phase-contrast microscope.
Statistical analysis
All data were derived from three independent experiments and are expressed as mean±standard deviation. Student's t-test or ANOVA was performed for statistical analysis. p-Value <0.05 was considered statistically significant.
Results
The expression of miR-21 was elevated in bladder cancer
By using quantitative RT-PCR, the relative levels of miR-21 to U6 RNA in the tumor tissue and paired adjacent noncancerous tissue from bladder cancer patients (n=3) who underwent radical cystectomy were determined and normalized to those of the normal bladder mucosa from patients (n=3) who had enucleation of the prostate due to prostatic hyperplasia (Fig. 1). The expression of miR-21 in primary bladder tumor tissue was considerably elevated, ∼3–4-fold compared with that of the adjacent noncancerous tissue and that of the normal bladder mucosa (ANOVA, p<0.05).

Expression of miR-21 in different bladder tissue samples was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR). The level of miR-21 in bladder tumor was compared with that in the paired adjacent noncancerous tissue and to that in the normal bladder mucosa (ANOVA, p<0.05).
Knockdown of miR-21 decreased cell proliferation in bladder cancer cell lines
The miR-21 levels in bladder cancer cell line J82 and RT112 cells transfected with the miR-21 ASO were knocked down by more than 70% compared with those in cells treated with the control ASO (Fig. 2A). At 72 h after the transfection, the miR-21 ASO-treated wells had significantly less viable cells than did the control ASO-treated wells (t-test, p<0.05), as measured by MTT assay (Fig. 2B). The reduction of viable cells upon miR-21 ASO treatment could be attributed to either cell apoptosis or alteration in cell proliferation. Thus, we performed mechanism studies to answer the question. Flow cytometric analysis with cells dual labeled by Annexin V-PE and 7-AAD showed that downregulation of miR-21 apparently had no effects on apoptosis of J82 and RT112 cells (Fig. 3). Colony formation assay showed that knockdown of miR-21 significantly decreased the numbers of J82 and RT112 colonies that formed on the plate compared with the control (t-test, p<0.05) (Fig. 4A, B). We then analyzed the cell cycle profiles of cells treated with miR-21 ASO and control ASO, respectively, using flow cytometry. Results showed that knockdown of miR-21 led to a higher percentage of cells in the G1 phase and, in the meantime, less cells in the S phase relative to the control in both J82 and RT112 cell line models (t-test, p<0.05) (Fig. 4C). This implicates that downregulation of miR-21 might slow down the proliferation of bladder cancer cells by arresting cells at the G1 phase.
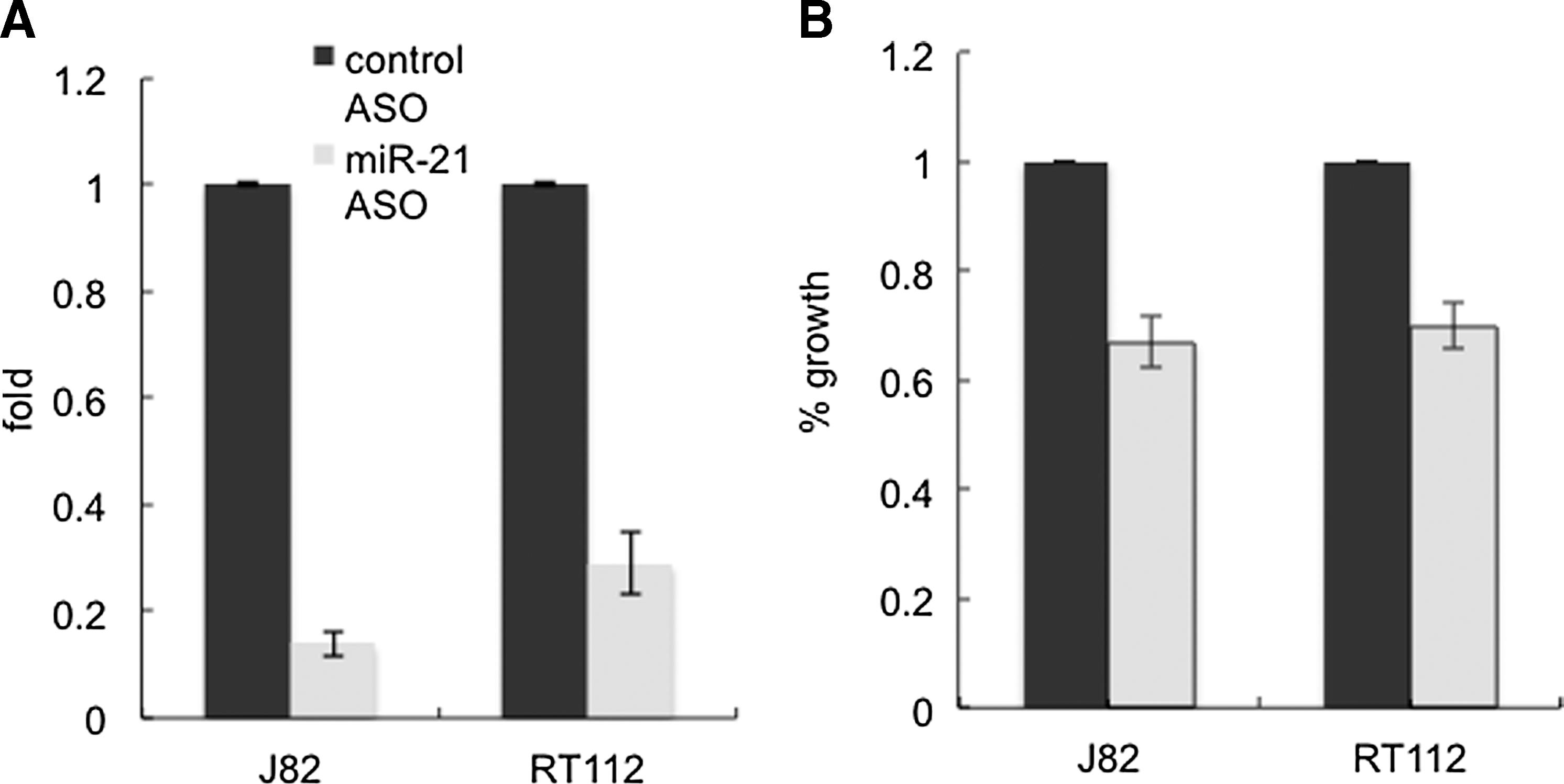
Knockdown of miR-21 suppressed growth of bladder cancer cells.

Knockdown of miR-21 did not cause cell apoptosis in bladder cancer cells. J82 cells or RT112 cells were transfected with control ASO or miR-21 ASO. After 48 h, cells were dual labeled by 7-AAD and Annexin V-PE, followed by flow cytometry analysis.
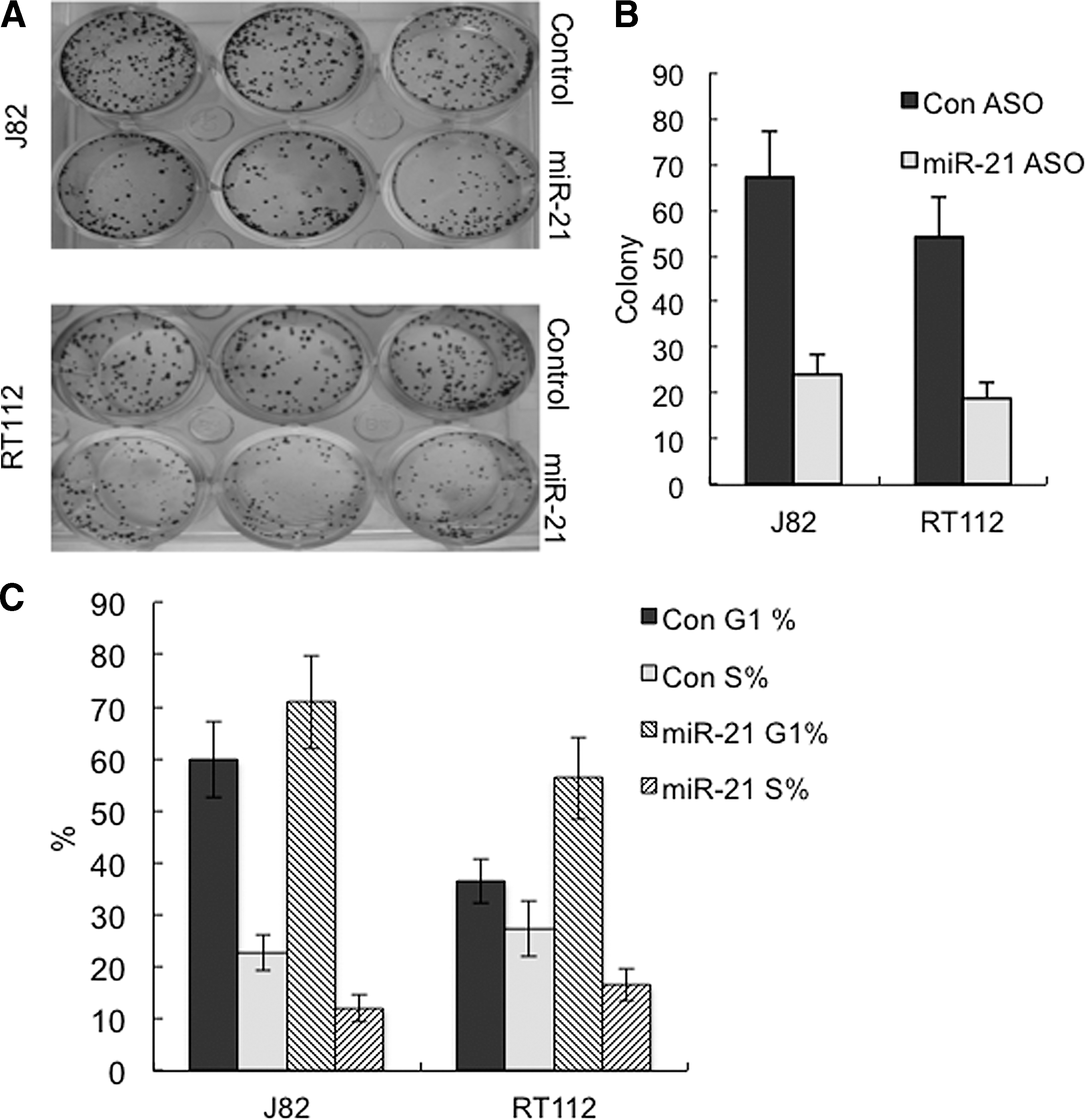
Downregulation of miR-21 reduced the colony formation of bladder cancer cells.
Knockdown of miR-21 suppresses the migration of bladder cancer cells
Wound healing assay showed slower closure of the wound of J82 cells transfected with miR-21 ASO than that of J82 cells transfected with control ASO (Fig. 5A). Transwell assay further showed that knockdown of miR-21 significantly decreased the number of J82 cells that migrated into the bottom surface of the membrane in the transwell chamber compared with the control (t-test, p<0.01) (Fig. 5B).
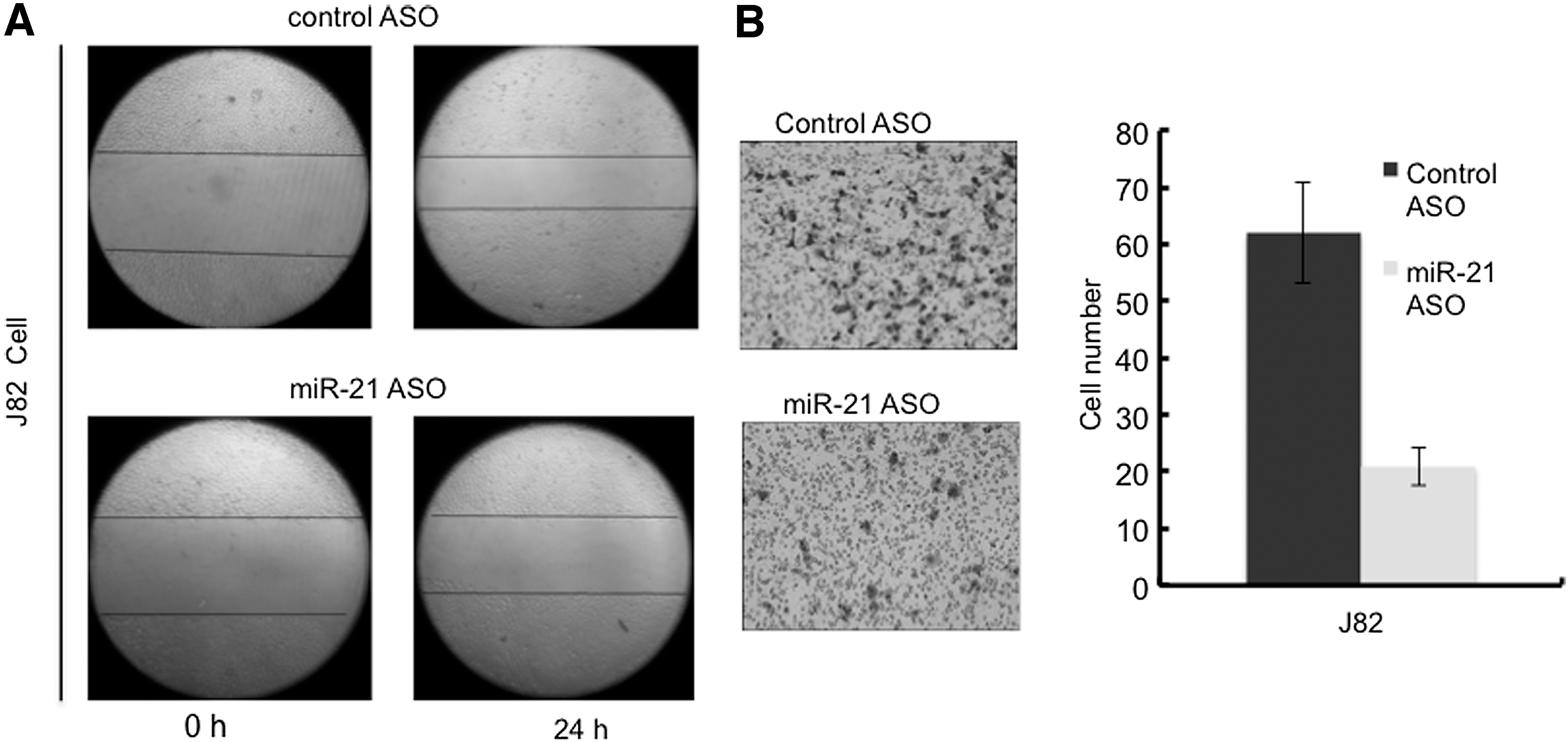
Knockdown of miR-21 decreased migration of bladder cancer cells.
Knockdown of miR-21 elevated the levels of PTEN and p53 phosphorylation at Ser46
PTEN has been demonstrated as a target gene of miR-21 (Meng et al., 2007), and expression of PTEN is inversely correlated with the miR-21 level in many cancer types (Kumarswamy et al., 2011). Western blot analysis showed that downregulation of miR-21 led to the increased expression of PTEN in bladder cancer lines (Fig. 6). We found that neither p53 level nor p53 phosphorylation at Ser15 had appreciable changes in response to the miR-21 ASO treatment. In contrast, we observed that phosphorylation of p53 at Ser46 increased when miR-21 was knocked down by the ASO (Fig. 6A, B). We also examined the acetylation state of p53 when miR-21 was decreased. The whole level of p53 acetylation was determined by pulling down the p53 protein and probing it with monoclonal antibody against acetylated lysine. The result showed that the total amount of acetylation of p53 had no notable alteration in response to miR-21 downregulation (Fig. 6C).
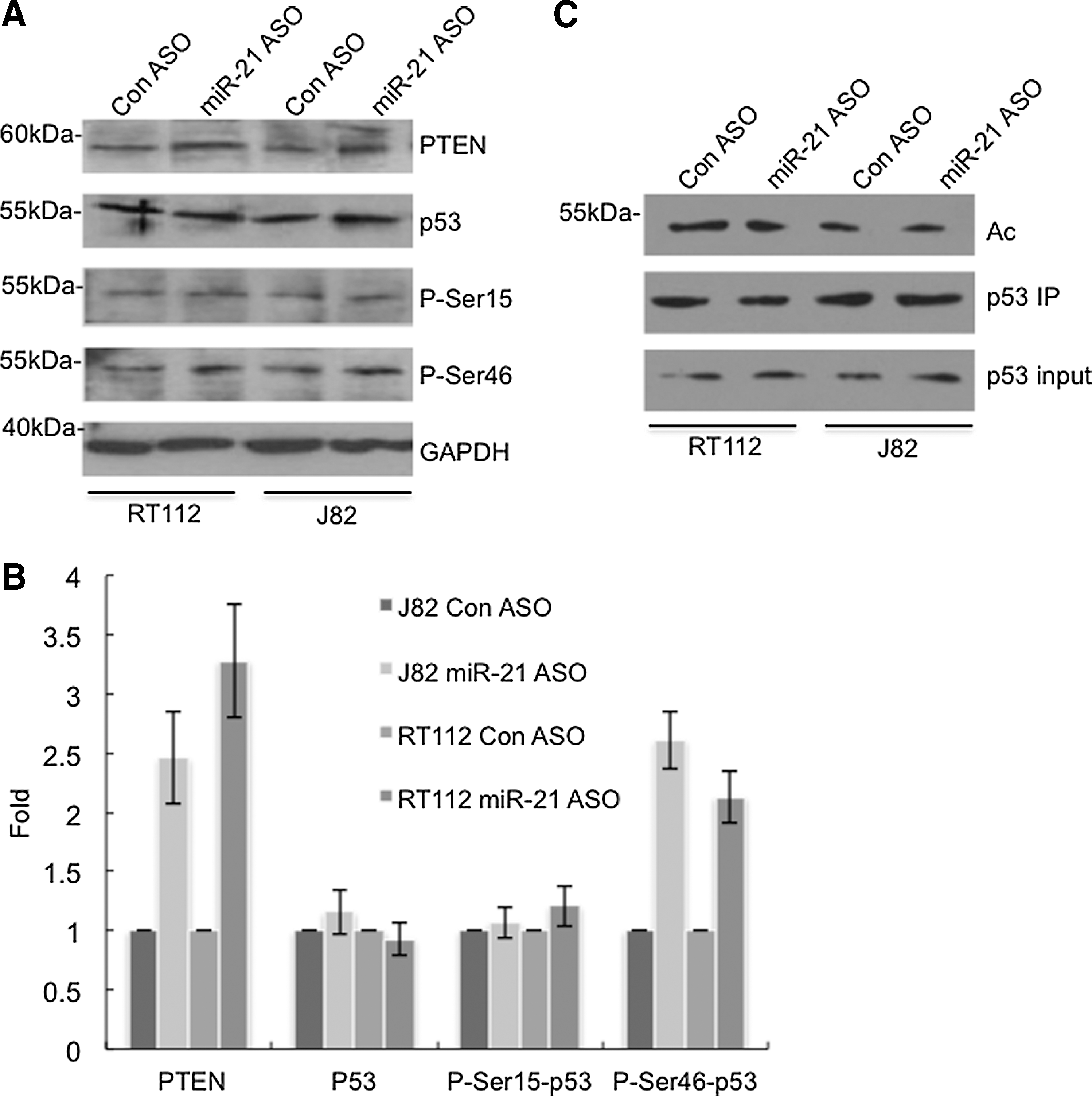
Knockdown of miR-21 led to the elevation of PTEN and phosphorylation of p53 at Ser46 in bladder cancer cells.
Discussion
Although miR-21 has been reported to be markedly upregulated in bladder tumor tissue (Dyrskjot et al., 2009; Neely et al., 2010) and associated with high-grade invasive tumor and poor prognosis (Catto et al., 2009), little was known about its oncogenic mechanism in bladder cancer. In the present study, with the tumor tissue from patients with bladder cancer (n=3) and the normal bladder mucosa from patients who underwent enucleation of the prostate due to prostatic hyperplasia (n=3), we observed that the level of miR-21 of the bladder tumor (Fig. 1) obviously increased compared with that of the normal mucosa, which is in agreement with the previous reports. Results from our loss-of-function studies support the oncogenic role of miR-21 in bladder cancer through regulating cell proliferation and migration. Downregulation of miR-21 did not cause cell apoptosis in bladder cancer cell lines, J82 and RT112 (Fig. 3), but led to the cell cycle arrest at the G1 phase (Fig. 4C). This might explain the slower growth rate and fewer colonies under miR-21 ASO treatment as analyzed by MTT assay (Fig. 2) and colony formation assay (Fig. 4A, B), respectively. Regarding the minimum effects of miR-21 knockdown on cell apoptosis in this study, we speculate that downregulation of miR-21 may not be sufficient to initiate the apoptotic program in bladder cancer cells or that other molecules compensated the loss of miR-21 in the models.
We examined the levels of PTEN and p53 in response to knockdown of miR-21. PTEN is an important tumor suppressor involved in cell growth, adhesion, and migration (Muller et al., 2011; Song et al., 2012; Mak, 2014). Mutation or deactivation of PTEN has been found in many cancer types (Hollander et al., 2011; Muller and Vousden, 2014). Typically, PTEN negatively regulates the PI3K/Akt signaling pathway to play a tumor-suppressive role (Seront et al., 2013). Our data showed that the PTEN level was significantly elevated upon miR-21 knockdown, implicating that miR-21 might promote tumor development in bladder cancer by targeting the tumor suppressor, PTEN. Our observation is consistent with the previous finding that PTEN is a target gene of miR-21(Xu et al., 2014).
The tumor suppressor, p53, plays a crucial role in biological processes, including cell cycle progression, apoptosis, and senescence (Rufini et al., 2013). Activation of p53 by phosphorylation at Ser15 is found to be preferentially associated with the cellular response to DNA damages (Loughery et al., 2014). In the present study, we did not see any appreciable change in the level of p53 or p53 phosphorylation at Ser15 when miR-21 was knocked down. Interestingly, we saw the increase of p53 phosphorylation at Ser46 along with the downregulation of miR-21 (Fig. 6). Phosphorylation of p53 at Ser46 has been shown to promote proapoptotic genes in several cancer types (Smeenk et al., 2011; Dashzeveg et al., 2014). However, in this study, knockdown of miR-21 in bladder cancer cells did not lead to apoptosis of J82 and RT112 cells (Fig. 3). It has been well established that acetylation is required for the full activation of p53 and in coordination with phosphorylation to regulate p53 activity (Tang et al., 2008; DeHart et al., 2014). Thus, we further examined the acetylation state of p53 protein. Our results showed that the whole level of p53 acetylation seemingly did not change when the level of miR-21 decreased. We reason that either the unique modification pattern of p53 protein preferentially triggered the cell cycle inhibition, but not the gene transcription program for apoptosis, or the cell cycle arrest was p53 independent as reported elsewhere (Hill et al., 2014). As miR-21 has multiple target genes (Buscaglia and Li, 2011), it is possible that derepression of certain cell cycle inhibitors upon the decrease of miR-21 directly caused the cells being arrested at the G1 phase without the involvement of p53.Our study just preliminarily explored the molecules possibly involved in the miR-21 regulatory network in bladder cancer, and many details are waiting to be investigated in future studies.
Conclusions
Our loss-of-function studies have shown that miR-21 plays a role in cell proliferation and migration in bladder cancer. The cross talk between miR-21 and the tumor suppressors, PTEN and p53, has also been suggested. Further studies are anticipated to uncover the signaling pathways and genes involved in the miR-21-PTEN/p53 axis.
Footnotes
Acknowledgments
This study was supported by grants from Tianjin Natural Science Foundation to L.L. (project No. 11JCYBJC13900), Science and Technology Innovation Fund of Tianjin Institute of Urology to L.M. (project No. MNYB201504), the anticancer key program of Tianjin Natural Science Foundation to H.R. (project No. 12zcdzsy16900), and National Natural Science Foundation of China to S.E. (project No. 81402095).
Disclosure Statement
No competing financial interests exist.