
Abstract
Ankylosing spondylitis (AS) is a chronic inflammatory rheumatic disease strongly associated with HLA-B*27, an major histocompatibility complex (MHC) molecule that presents peptide antigen to T cells. Previously, regulatory B cells were found to suppress T cell-mediated autoimmunity induction and chronic inflammation, partially through interleukin (IL)-10 production. Here, we examined the role of regulatory B cells in AS pathogenesis. Apheresis samples from HLA-B*27-positive AS patients and non-AS healthy controls were collected. We found that although AS patients and non-AS controls presented similar frequencies of CD24+CD38+ B cells, compared to non-AS controls, those from AS patients produced less IL-10 under ex vivo condition and after CD40 and B-cell receptor (BCR) stimulation. Purified T cell-B cell cocultures showed that compared to non-AS controls, CD24+CD38+ B cells from AS patients were defective at suppressing naive and memory CD8+ T cell activation. The suppression of memory CD8+ T cells in non-AS controls appeared to be mediated by IL-10, since the addition of IL-10 mAb suppressed CD24+CD38+ B cell-mediated downregulation of proinflammatory cytokine production and proliferation. To rescue the defect in AS patients, CD24+CD38+ B cells were pretreated by CD40 and BCR stimulation, which enhanced CD24+CD38+ B cell-mediated memory CD8+ T cell suppression. Together, our data discovered a regulatory B cell defect in AS patients.
Introduction
A
Since HLA-B*27 allele is a strong positive risk factor (Taurog, 2010), the prevalent opinion on AS pathogenesis explains that exogenous antigens presented by HLA-B*27 could activate a T cell-mediated autoimmune response through molecular mimicry; the activated CD8+ T cells then attack auto-antigens in the joint tissue to exacerbate chronic inflammation (Fiorillo et al., 2000; Tam et al., 2010). Inline with this hypothesis, multiple immune-related loci were associated with risk for AS by genotyping studies (Lin et al., 2011; International Genetics of Ankylosing Spondylitis Consortium et al., 2013). However, although 80% AS patients are carriers of HLA-B*27, only 1–5% of all HLA-B*27 carriers have this disease (Brown et al., 1997, 2000). Moreover, multiple studies have revealed an association between the mutations and expressions of immunoregulatory genes (Reveille et al., 2010; Evans et al., 2011; Lin et al., 2011; International Genetics of Ankylosing Spondylitis Consortium et al., 2013), suggesting that immune regulatory mechanisms are in place to prevent or exacerbate disease development.
The role of regulatory B cells in controlling excessive inflammation and preventing autoimmune induction has been demonstrated in mouse models and human diseases (Mauri and Blair, 2010). Adoptive transfer of CD1dhiCD5+ B cells substantially reduced T cell-mediated inflammation in mice autoimmune models (Yanaba et al., 2008, 2009). CD24+CD38+ B cells suppressed virus-specific adaptive T cell responses in humans (Das et al., 2012). The production of interleukin (IL)-10 was thought to be the primary mechanism by which regulatory B cells exerted suppression, although other mechanisms, including immunosuppressive antigen presentation, were also proposed (Mauri et al., 2003; Tedder and Leonard, 2014). The development of regulatory B cells in autoimmunity and inflammatory bowel disease mouse models is dependent on CD40 signaling (Fillatreau et al., 2002; Mizoguchi et al., 2002). Engagement of CD40 on B cells could directly cause IL-10 production (Duddy et al., 2007). Interestingly, in several autoimmune diseases, including systemic lupus erythematosus (SLE) and immune thrombocytopenia, the CD24+CD38+ B cells lacked the regulatory capacity observed in their counterparts in healthy subjects (Blair et al., 2010; Li et al., 2012), implicating a role of regulatory B cell defect in autoimmunity. Here, we studied the functions of B cell-mediated suppression in AS.
Materials and Methods
Patients
A total of 30 subjects were recruited, including 15 AS patients (13 men, 2 women) between 27 and 60 years of age, and 15 non-AS healthy controls (13 men, 2 women) between 25 and 65 years of age (Table 1). All AS patients were diagnosed AS according to the modified New York criteria (van der Linden et al., 1984). All AS cases and controls were HLA-B27-positive. No treatment with nonsteroidal anti-inflammatory drug and/or disease-modifying antirheumatic drug was given before sample collection. Written informed consent was obtained from all cases with approval from the ethics committee of Qianfoshan Hospital, Shandong University. Peripheral blood mononuclear cells (PBMCs) were obtained from Ficoll centrifugation of apheresis samples collected at the time of diagnosis. For IL-10 production and B cell surface marker examination, fresh cells were used directly; for T cell-B cell culture, cells were cryopreserved at −80°C for <1 month, thawed, and rested at 37°C 5% CO2 overnight before use.
All continuous values were expressed and mean ± standard deviation.
AS, ankylosing spondylitis; N/A, not applicable.
Flow cytometry
For surface marker staining, PBMCs were suspended in FACS staining buffer (phosphate-buffered saline supplemented with 1% bovine serum albumin, 2 nM EDTA, and 0.1% sodium azide). Surface antibodies, including anti-human CD3, CD8, CD19, CD20, CD21, CD24, CD27, CD38 (BioLegend), CD45RA, CD45RO, CD62L, and/or IgD (BD), together with LIVE/DEAD Fixable Aqua (Invitrogen), were added for 30 min at 4°C. Cells were then washed twice with FACS staining buffer. For intracellular staining, surface-stained cells were incubated in CytoFix/CytoPerm (BD) for 20 min at 4°C, washed twice with 1× Perm/Wash (BD), incubated with anti-human IL-10, IFN-gamma, and/or TNF-alpha antibodies for 30 min at 4°C, and then washed twice. After staining, cells were fixed with 2% formaldehyde and were analyzed by BD FACSCanto II. For live cell sorting, PBMCs were incubated in complete culture media (RPMI 1640 supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine, and 10% heat-inactivated fetal calf serum; Sigma) with surface antibodies for 30 min at 4°C. Cells were then washed twice in complete culture media and were sorted in BD FACSAria.
Cell culture
All cell cultures were maintained at 106 cells per 1 mL complete culture media, at 37°C 5% CO2. If noted, anti-human CD40 and IgM (BioLegend) were added at 1 μg/mL per 106 B cells for stimulation. IL-10 production was measured by Luminex at 72 h. T cell+autologous B cell were cultured at 1:1 ratio. Live-sorted T cells were first pretreated with carboxyfluorescein succinimidyl ester (CFSE) before coincubation with B cells, with or without 10 μg/mL unlabeled IL-10 mAb (BD). After 72 h coincubation, T cells were negatively magnetic bead-purified by Human CD8 T cell Enrichment Kit (Stemcell), stimulated with anti-human CD3 and CD28 (BioLegend) monoclonal antibodies for 6 h, and analyzed by flow cytometry.
Statistical analysis
All datasets were first examined by D'Agostino-Pearson normality test, followed by parametric or nonparametric two-tailed tests accordingly, in Prism (GraphPad).
Results
Comparable proportions of CD24+CD38+ B cells in AS patients and non-AS controls
The composition of circulating B cells was examined (Fig. 1A). We found that the numbers of CD19+ B cells were comparable between healthy (non-AS) and patients (Fig. 1B). Within the B cell compartment, AS patients contained higher frequencies of CD20hiCD21lo activated B cells, IgD-CD27+ memory B cells, and CD19loCD20-CD27hi plasmablasts, while healthy patients contained higher frequencies of IgD+CD27− naive B cells (Fig. 1B). The frequencies of CD24+CD38+ B cells were comparable between the two groups.
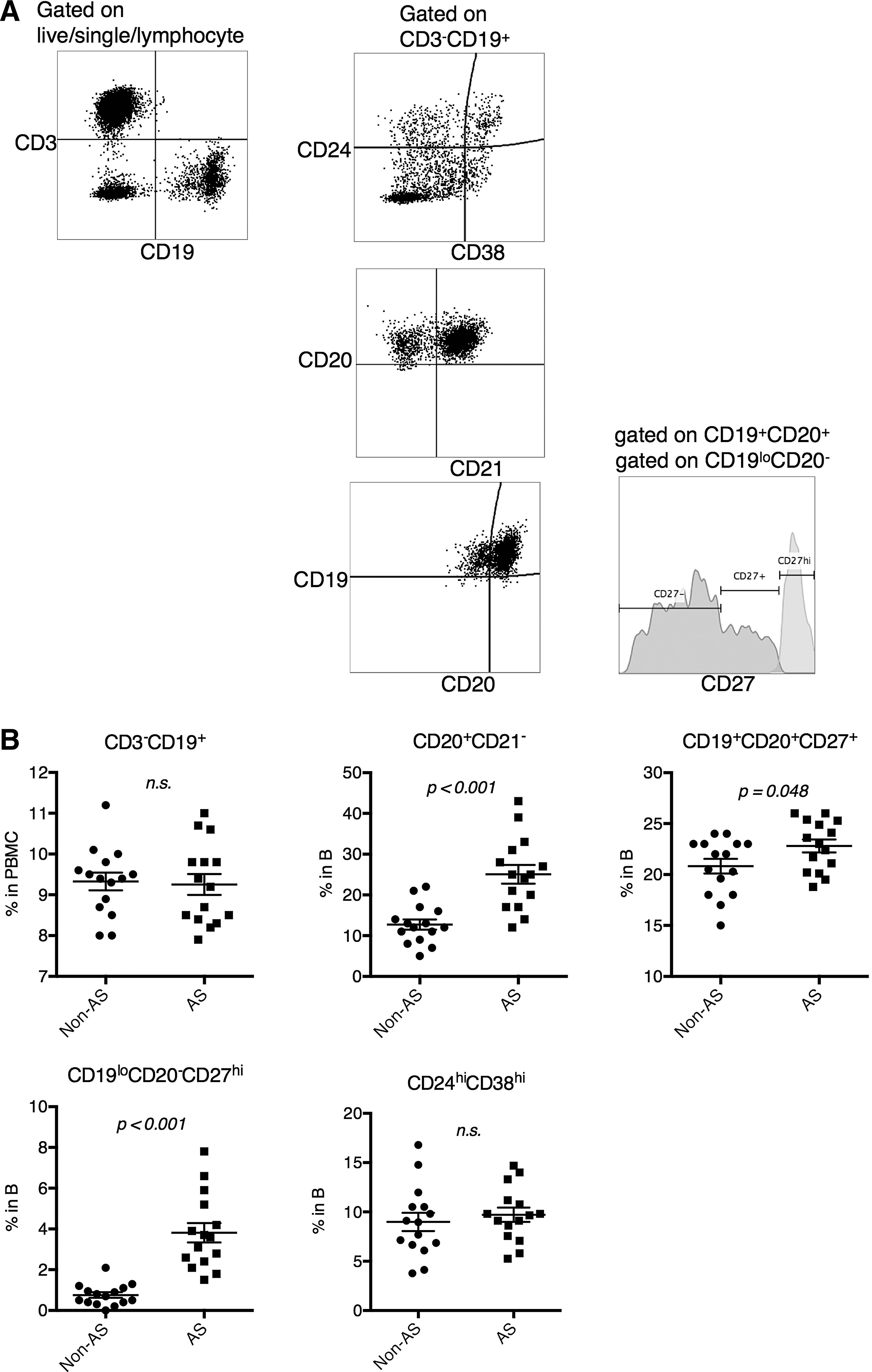
Peripheral B cell composition was altered in AS patients.
IL-10 production was concentrated within the CD24+CD38+ compartment
Both CD19loCD20−CD27hi plasmablasts and CD24+CD38+ B cells were ascribed with IL-10 producing functionality (Blair et al., 2010; Matsumoto et al., 2014). Here, we examined IL-10 production in AS. B cell subpopulations were live-sorted and cultured separately based on their surface marker expression. We were unable to recover sufficient numbers of plasmablasts from healthy subjects due to their low representation in peripheral blood. IL-10 expression was then measured by Luminex. We found that under directly ex vivo conditions, very little IL-10 was produced by any B cell subpopulations except CD24+CD38+ B cells from healthy subjects (Fig. 2A). After CD40+B-cell receptor (BCR) stimulation, the CD24+CD38+ B cells in healthy subjects produced significantly higher IL-10, than those from AS patients (Fig. 2B). In AS patients, CD24+CD38+ B cells produced similar amounts of IL-10 with CD20-CD27hi plasmablasts under ex vivo condition, and slightly higher amounts under CD40+BCR stimulation (Fig. 2A, B), when the cell number was equalized in each culture. Since CD24+CD38+ B cells were present in the peripheral blood at higher frequencies (Fig. 1B), we expected that CD24+CD38+ B cells represented the primary IL-10-producing cells in vivo. This was confirmed by flow cytometry staining (Fig. 2C), in which we found most IL-10-producing cells belonging to the CD24+CD38+ B cell subpopulation (Fig. 2D).
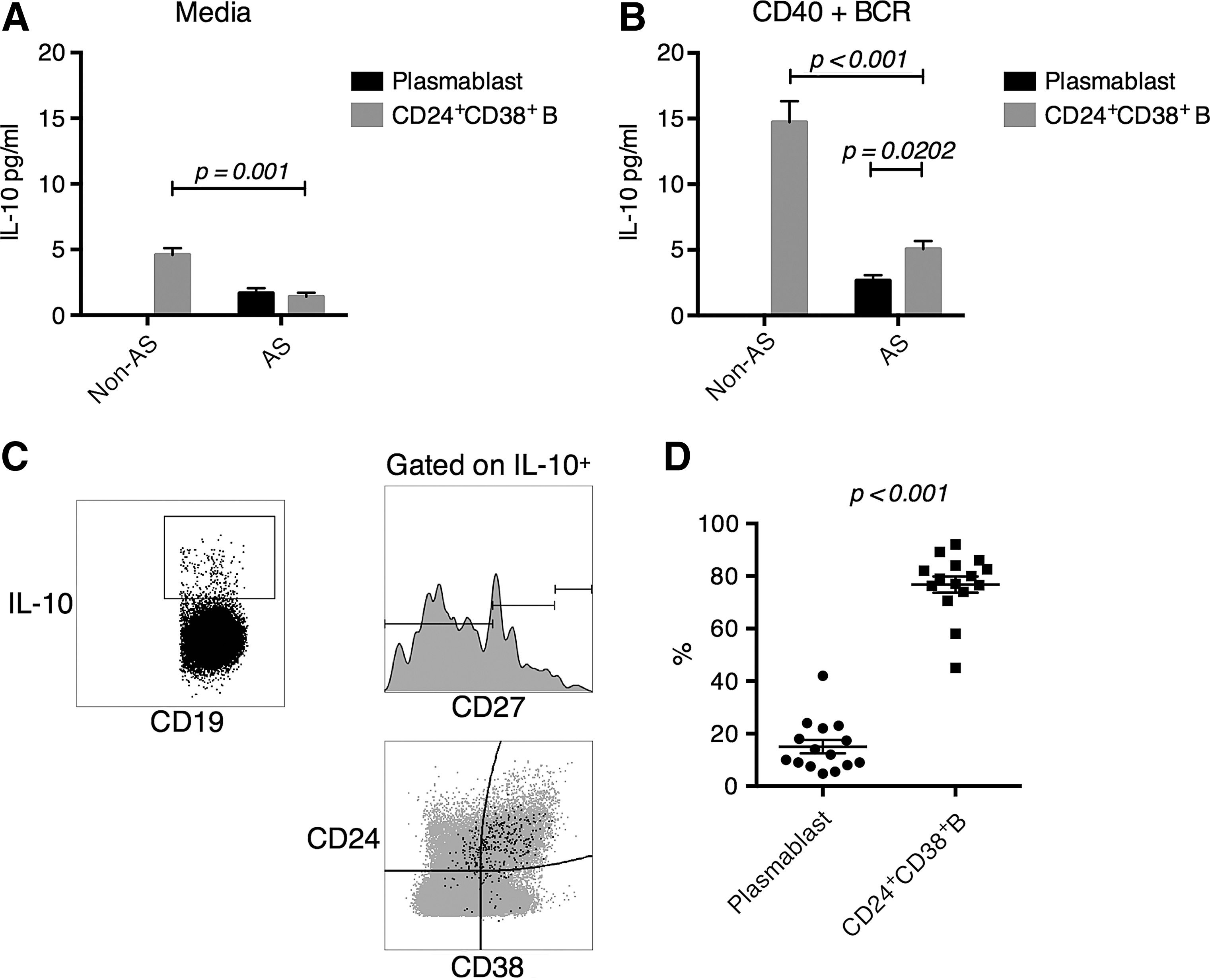
IL-10 expression was primarily mediated by CD24+CD38+ B cells in both non-AS and AS subjects, while in AS subjects the IL-10 production was reduced.
CD24+CD38+ B cells in healthy, but not AS, subjects suppressed naive CD8+ T cell activation
Previously, regulatory B cells were shown to suppress both CD4+ and CD8+ T cell antigen-specific inflammation (Liu et al., 2014). We focused on their effect on naive and memory CD8+ T cells since AS is thought to be primarily CD8+ T cell-mediated. T cells were first sorted into naive (CD45RA+CD62L+) and memory (CD45RO+) T cells (Fig. 3A) (Maldonado et al., 2003). Live-sorted naive CD8+ T cells were then cocultured with autologous B cell subgroups for 3 days, after which the naive CD8+ T cells were purified and restimulated (Fig. 3A). T cell activation was evaluated by IFN-gamma and TNF-alpha expression and CFSE staining (Fig. 3B). We found that CD24+CD38+ B cells from healthy subjects could potently suppress naive CD8+ T cell activation (Fig. 3C). The suppression did not seem to be primarily induced by IL-10, since depletion of IL-10 from the coculture by IL-10 mAb did not significantly revert the suppression. In AS patients, neither CD27hi plasmablasts nor CD24+CD38+ B cells suppressed naive CD8+ T cell activation (Fig. 3D).
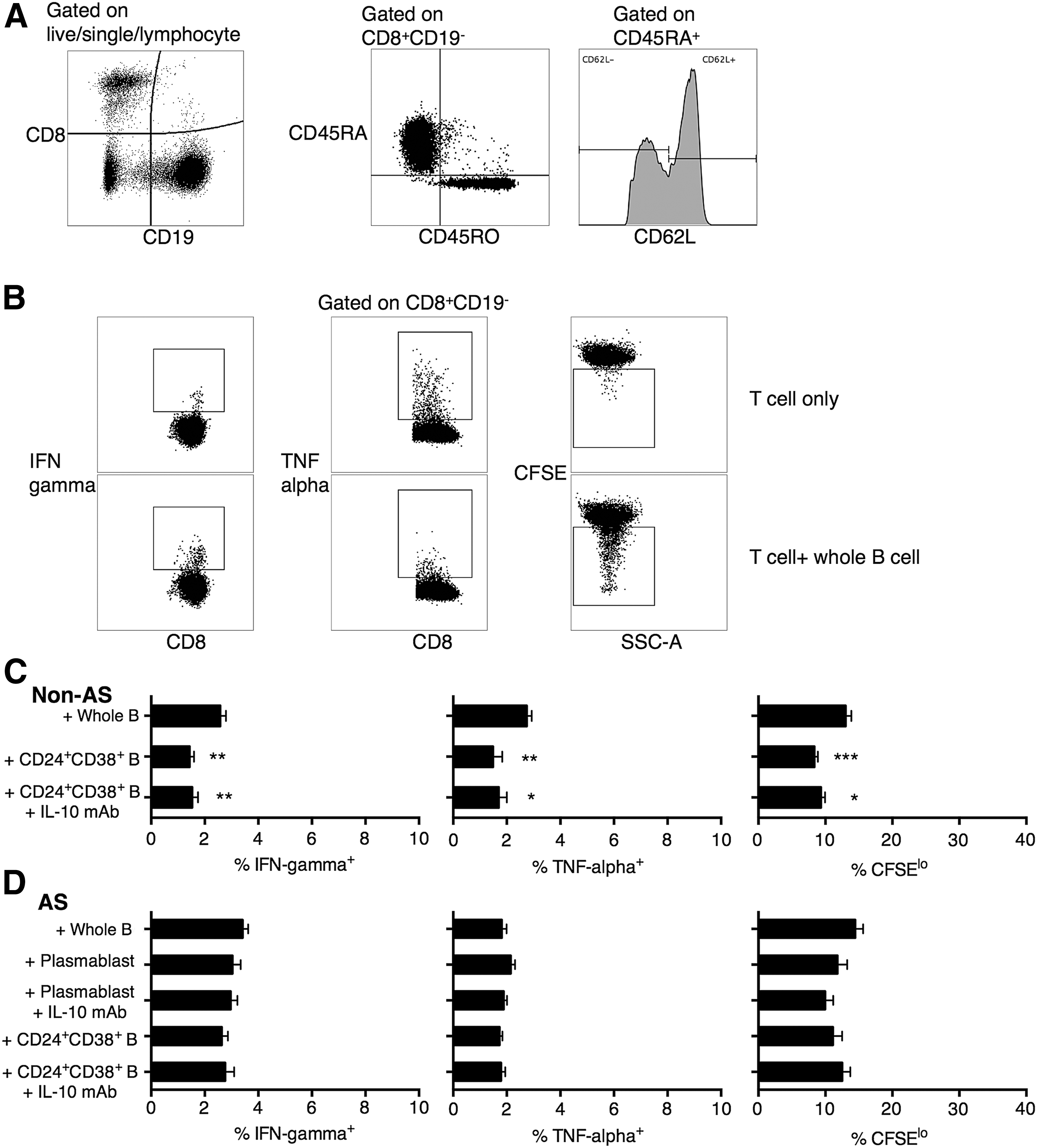
CD24+CD38+ B cells exerted regulatory function on naive T cells in non-AS subjects through IL-10-independent pathway, a function that was absent in AS subjects.
CD24+CD38+ B cells in healthy, but not AS, subjects suppressed memory CD8+ T cell activation, in part due to IL-10 expression
We next examined the suppression of memory CD8+ T cells by regulatory B cells. Again, we found that CD24+CD38+ B cells from healthy subjects could potently suppress memory CD8+ T cell activation, possibly through an IL-10-mediated pathway, since this time, depletion of IL-10 was shown to enhance CD8+ T cell IFN-gamma and TNF-alpha production, and partially enhance proliferation (Fig. 4A). Inline with this supposition, the level of suppression by CD24+CD38+ B cells in non-AS subjects was directly correlated with the level of IL-10 secretion, in terms of IFN-gamma and CFSE (Fig. 4C). Again, in AS patients, neither CD19loCD20-CD27hi plasmablasts nor CD24+CD38+ B cells suppressed memory CD8+ T cell activation, presumably due to the lack of IL-10 expression ex vivo (Fig. 4B).
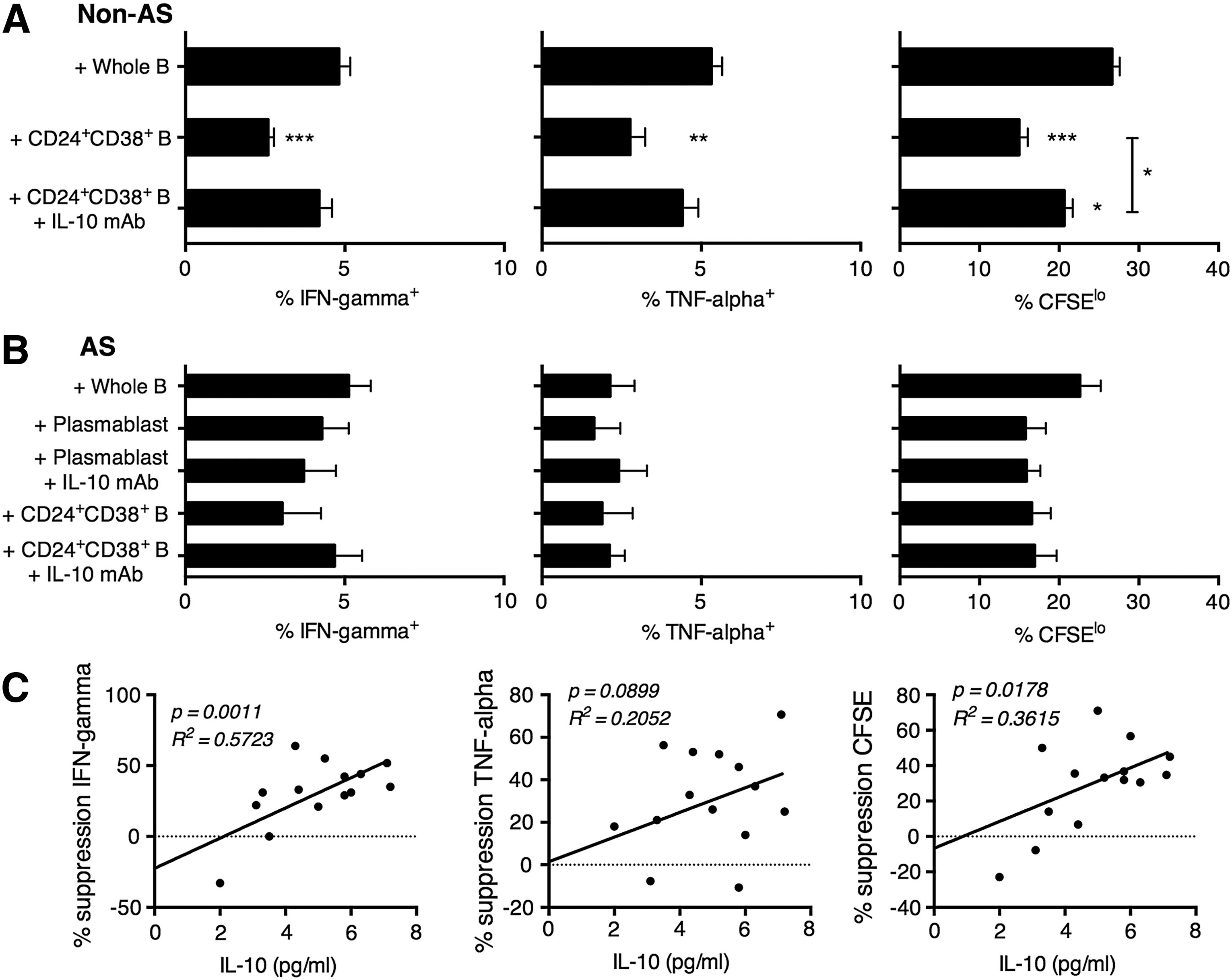
CD24+CD38+ B cells exerted regulatory function on memory T cells in non-AS subjects through IL-10-dependent pathway, a function that was absent in AS subjects. The percentage of IFN-gamma+, TNF-alpha+, and CFSElo memory (CD45RO+) T cells after coculture with autologous whole B cells, CD24+CD38+ B cells, or CD24+CD38+ B cells+anti-IL-10 monoclonal antibody (IL-10 mAb), in
In an attempt to rescue the suppressive effect of CD24+CD38+ B cells in AS patients, we pretreated the live-sorted CD24+CD38+ B cells with CD40+BCR stimulation to boost their IL-10 production. The autologous memory CD8+ T cells were then cocultured with stimulated CD24+CD38+ B cells, purified, and restimulated as previously described (Evans et al., 2011). We found that compared to ex vivo CD24+CD38+ B cells, CD40+BCR-pretreated CD24+CD38+ B cells significantly suppressed memory CD8+ T cell activation (Fig. 5).

Defective CD24+CD38+ B cell function in AS patients can be rescued by CD40+BCR stimulation. CD24+CD38+ B cells were pretreated without (media) or with anti-CD40+anti-BCR stimulation. The percentage of IFN-gamma+, TNF-alpha+, and CFSElo memory (CD45RO+) T cells after coculture with autologous CD24+CD38+ B cells were then examined. Mean ± SEM Mann–Whitney test.
Discussion
The fact that only a minority of HLA-B*27 carriers will develop AS indicates that immunoregulatory mechanisms are possibly in place to prevent the inflammation leading to AS pathogenesis. Indeed, defective suppressive functions by regulatory B cell subsets, including CD1dhiCD5+ mouse B cells, CD24+CD38+ human B cells, and classical memory B cells, have been observed in several chronic inflammation diseases (Yanaba et al., 2008; Blair et al., 2010; Das et al., 2012; Li et al., 2012; Liu et al., 2014). Here, we observed that AS patients also presented defective regulatory B cell function. Phenotypically, the peripheral blood B cell population in AS patients contained significantly elevated frequencies of activated B cells, memory B cells, and plasmablasts, compared with non-AS controls, while the no significant differences were observed in the CD24+CD38+ population. Functionally, we observed that the IL-10-producing population was concentrated in CD24+CD38+ B cells in AS subjects, and was much reduced compared with that in non-AS subjects, both under ex vivo condition and after CD40 and BCR stimulation. Previously, upregulation of CD24+CD38+ B cells was observed in SLE but not osteoarthritis patients (Blair et al., 2010). Here, we observed that although AS patients did not present an upregulation of CD24+CD38+ B cells, their CD24+CD38+ B cell population presented reduced IL-10-producing function, which can be partially but not fully rescued by CD40 and BCR stimulation.
Previous studies on regulatory B cell suppression examined their effect on total T cells. Since naive T cells and memory T cells require different survival, activation and regulation signals (Stemberger et al., 2007), we examined the effect of CD24+CD38+ B cells on naive and memory CD8+ T cells separately. We found that in non-AS controls, CD24+CD38+ B cells suppressed naive CD8+ T cell proinflammatory cytokine production and proliferation, but addition of IL-10 mAb did not revert the CD24+CD38+ B cell-mediated suppression of naive CD8+ T cells, which contrasted with previous reports showing a partial restoration of whole CD8+ T cell inflammation by IL-10 removal (Blair et al., 2010; Liu et al., 2014). In contrast, CD24+CD38+ B cells suppressed memory CD8+ T cell proinflammatory cytokine production and proliferation, and addition of IL-10 mAb reverted the CD24+CD38+ B cell-mediated suppression of memory CD8+ T cells. These comparisons revealed that CD24+CD38+ B cells acted by other mechanisms to suppress naive CD8+ T cell activation. CD24+CD38+ B cell was previously identified as an immature/transitional population (Sims et al., 2005; Plebani et al., 2007; Palanichamy et al., 2009). Whether these B cells lacked the proper antigen presentation and costimulation machinery to induce naive T cell activation is unknown. In terms of memory CD8+ T cell activation, however, IL-10 secretion appeared to be the primary mode of action. The lack of either plasmablast- or CD24+CD38+ B cell-mediated suppression of naive or memory CD8+ T cells in AS patients provided a potential mechanism of inflammation induction during AS pathogenesis.
The observation that regulatory B cell function in AS patients could be rescued by CD40+BCR stimulation of CD24+CD38+ B cells suggested a possible therapeutic strategy. In future studies, the effects of suppressing CD8+ T cell inflammation in AS patients need to be assessed. Previously, regulatory B cells were seen to promote Treg differentiation by increasing Foxp3 and CTLA4 expression (Kessel et al., 2012), and inhibit Th1/Th17 differentiation (Lampropoulou et al., 2008; Carter et al., 2011; Wang et al., 2014). Given that mechanisms mediated by CD4+ T cells, including IL-17 production and KIR2DL2+ T cell upregulation, were associated with AS pathogenesis (Bowness et al., 2011; Baeten et al., 2013), these other effects of regulatory B cell function and their impact on AS pathogenesis need to be examined.
Footnotes
Acknowledgments
The work was supported by Shandong Provincial Natural Science Foundation, China (No. ZR2014HM049), and National Natural Science Foundation of China (No. 81301533).
Disclosure Statement
No competing financial interests exist.