
Abstract
Recently, post-transcriptional gene regulation by microRNAs (miRNAs) has been reported to play a key role during ovary development and differentiation. However, there are no published studies identifying miRNA profiles of human ovarian tissues directly using next-generation sequencing technology. In the human ovary, a total of 762 known and 21 novel human miRNAs were detected, indicating that human ovaries have a complex population of small RNAs. To confirm the miRNA profile in human ovaries, quantitative real-time polymerase chain reaction was used to validate the expression of known miRNAs and novel miRNAs. The potential regulating roles of miRNA in physiological function of ovaries were analyzed by gene ontology and Kyoto encyclopedia of genes and genomes pathway annotation, and several important processes were identified to be targeted by the most abundantly expressed miRNAs, for example, antral ovarian follicle growth, ovarian follicle rupture, and fertilization. Our current findings extend the knowledge of the regulatory role of miRNAs and their targeted processes in human ovaries, suggesting that miRNAs play important roles in development and physiological function of ovaries. In this study, we provide a useful resource for further research of the regulatory role of miRNAs in the ovaries, which may also provide novel candidates for molecular biomarkers or treatment targets in the research of female infertility.
Introduction
T
Recently, post-transcriptional gene regulation by microRNAs (miRNAs) has been reported to play a key role during ovary development and differentiation (Hawkins and Matzuk, 2010; Li et al., 2015). miRNAs are small noncoding RNAs, average 21 nucleotides in size, and regulate gene expression through transcriptional repression or degradation based on direct base-pairing interactions with targeted mRNAs at the post-transcriptional level (Azuma-Mukai et al., 2008). Most miRNAs are evolutionary, conserved in a large number of species, and function in most tissue development and differentiation (Azuma-Mukai et al., 2008; Carletti and Christenson, 2009; Sorensen et al., 2014).
The role of miRNAs in ovarian function has been investigated through Dicer knockout mouse models (Bernstein et al., 2003). Dicer is an important RNase III enzyme required for mature miRNA production, and general knockout of Dicer in mice resulted in embryonic lethality (Bernstein et al., 2003). Using conditional knockout mouse models of Dicer in ovarian granulose cells, these mice showed a reduction in ovarian weight and ovulation rates, abnormal estrous cycles, shorter estrus, and longer metestrus, paratubal cyst, and even infertility (Lei et al., 2010). Oocytes with Dicer-specific knockout exhibited an impaired ability to extrude a polar body, multiple spindles, and chromatin condensation defects (Liu et al., 2010; Yuan et al., 2014). These results suggested that miRNAs are necessary for the maintenance of ovarian function and even fertility.
Mammalian ovaries exhibit spatiotemporal mRNA expression pattern as well as miRNAs (Lei et al., 2010; Tong et al., 2014; Zhang et al., 2014). Moreover, miRNAs are the most abundant class of small RNAs in the ovary in different mammalian species (Carletti and Christenson, 2009; Ahn et al., 2010; Chan and Ruohola-Baker, 2010; Hackl et al., 2011). The miRNA expression profile in mouse, cattle, rat, and porcine was identified by microarray, high-throughput polymerase chain reaction (PCR), and next-generation sequencing techniques (Ahn et al., 2010; Hackl et al., 2011; Miretti et al., 2011; Podolska et al., 2011; Lin et al., 2015). These results suggested that several miRNAs, for example, the let-7 family, miR-21, miR-99a, and miR-199b, expressed abundantly in these species and bioinformatic analysis revealed that these miRNAs might be involved in regulation of several biological processes (Hackl et al., 2011; Miretti et al., 2011; Podolska et al., 2011; Shi et al., 2014; Wagner et al., 2014; Lin et al., 2015).
For example, the expression of miRNAs in the let-7 family was required for mammalian developmental timing and tumor suppressor function (Lee et al., 2015). MiR-21 plays crucial roles in inflammatory injury and carcinogenesis (Liu et al., 2013; Yin et al., 2015). MiR-99a could inhibit cell proliferation and regulate tumor progression and stem-like properties in cholangiocarcinoma (Huang et al., 2015; Lin et al., 2015). MiR-199b-5p could enhance the suppression on cell migration and could also participate in human erythropoiesis (Fang et al., 2013; Li et al., 2014).
Although the miRNA expression and function have been detected in mice and other mammalian models, the miRNA expression profile of human ovarian tissues was not documented in detail. In this study, we determined the miRNA expression profile of normal human ovaries and identified the abundantly expressed miRNAs and novel miRNAs using next-generation sequencing (NGS) technology. Moreover, bioinformatic analysis, gene ontology (GO) annotation, and pathway enrichment of the potential miRNA target genes for the abundantly expressed miRNAs indicated that many important processes related with ovarian function were enriched, such as antral ovarian follicle growth, ovarian follicle rupture, and fertilization. Interestingly, several similar biological processes were highlighted for the novel miRNA targets, for example, the MAPK signaling pathway. Our results suggested that miRNA in human ovaries plays important role in oogenesis, folliculogenesis, and other ovarian functions and this offers new insights for the treatment of reproductive and metabolic disorders associated with ovaries.
Materials and Methods
Ovary sample collection
The ovary tissues used for NGS were collected from three cadavers (aged 22, 27, and 31 years) from Anhui Provincial Hospital Affiliated with Anhui Medical University, Hefei, China. These samples with normal folliculogenesis enrolled in this study after histological examination. Detailed basic information of the three subjects is listed in Supplementary Table S1 (Supplementary Data are available online at
Small library construction and sequencing
The small RNA sequencing for human ovaries was completed at BGI-Shenzhen (Tong et al., 2014; Xu et al., 2015). Total RNAs were extracted from ovary samples using TRIzol reagent (Invitrogen). The samples from three subjects were pooled homogeneously into one RNA sample. Thus, the total RNA samples were subjected to 15% (W/V) denaturing polyacrylamide gel electrophoresis and 18–22 nt small RNA fragments were isolated. These isolated small RNAs were ligated to the adaptors: 5′ adaptor-GTTCAGAGTTCTACAGTCCG-ACGATC, 3′ adaptor-TCGTATGCCGTCTTCTGCTTG, and then these RNAs were transcribed by reverse transcription PCR. After purification, the reverse transcription PCR (RT-PCR) products were sequenced by the Illumina Hiseq 2000 (Illumina) according to Illumina's protocol.
Computational analysis of sequencing data
The NGS data of human ovary were analyzed by our previously published tools, CPSS (Zhang et al., 2012). Briefly, the adaptor sequences, low quality reads, and contaminated reads were first removed, and the remaining sequences were counted as the tags. These tags were mapped to the human genome using SOAP2.0 (Li et al., 2009), and all the mapped tags were matched into several RNA databases, such as miRbase (Van Peer et al., 2014), Rfam (Burge et al., 2013), Genebank nonoding RNA database (Karolchik et al., 2014), piRNA database (Li et al., 2009), or repeats database (Tarailo-Graovac and Chen, 2009). Only the tags that matched with miRbase were classified as known miRNAs and submitted into further analysis. The tags were classified as unclassified tags if they were not assigned to any of the above databases, which were used for the prediction of novel miRNAs in human ovaries.
Prediction of novel miRNAs in human ovaries
To avoid false positive by discarding the candidates with low abundance, the unclassified tags with 45 reads were processed for novel miRNA prediction. MiRDeep (An et al., 2013) and Mireap (
Bioinformatic analyses for known miRNAs in human ovaries
The bioinformatic analyses for known miRNAs were processed as our previous reports. In this study, the putative targets of the most abundantly known miRNAs in human ovaries were predicted by Targetscan (Lewis et al., 2003), MicroCosm (
miRNA expression detection by quantitative real-time PCR
According to our previous reports (Zhang et al., 2012, 2014), miRNA quantification was detected by real-time PCR by using the Applied Biosystems StepOne™ Real-Time PCR System (Applied Biosystems) and an SYBR premix Ex Taq™ II kit (Takara) with the primers listed in the Supplementary Table S2. The snRNA level of U6 was used as an internal reference. In this study, the PCR reactions were performed at 95°C for 30 s, followed by 40 cycles at 95°C for 5 s and 60°C for 31 s.
Statistical analysis
In this study, the PCR experiments were run in triplicate for each of the three samples. Quantitative data from real-time PCR were compared using Student's t-tests. p < 0.05 was considered significant.
Results
Overview of small RNA sequencing data
In this study, we obtained a total of 9,132,054 raw reads from human ovaries using Solexa NGS technology. After removal of 5′ and 3′ adaptor sequences, contaminations, and low quality reads, 8,403,080 clean reads, representing 283,647 unique tags, remained (Table 1). The majority size of these unique tags was 22 nt, varying between 18 and 26 (Supplementary Fig. S1). For detecting the miRNA expression profile from these reads, all the tags were matched into miRbase (V14.1). Thus, 762 miRNAs corresponding to 5,672,362 reads in human ovaries were identified as known miRNAs. In this study, after identification of known miRNAs, the rest of the reads were matched into other small RNA databases, including rRNAs, repeats, and snRNAs. The chromosome locations of the clean reads were analyzed and the distributions are shown in Supplementary Figure S2. In human ovaries, most reads are located in chromosome 9, followed by chromosome 22, 21, and 16, respectively.
The expression and enzymatic modification of known miRNAs
To identify known miRNA expression profile in human ovaries, the counts of each type of miRNA were normalized (normalized counts are displayed as reads per million) and 10 miRNAs with most abundant reads counts were listed (Table 2), indicating that they are highly expressed in human ovaries. To validate the miRNA expression detected by sequencing, 10 known miRNAs representing different expression levels were randomly chosen for quantitative real-time PCR. The expression pattern of these 10 miRNAs detected by quantitative real-time PCR was consistent with the results obtained from sequencing, and these results indicated that the miRNA expression identified by NGS technology was reliable (Fig. 1).
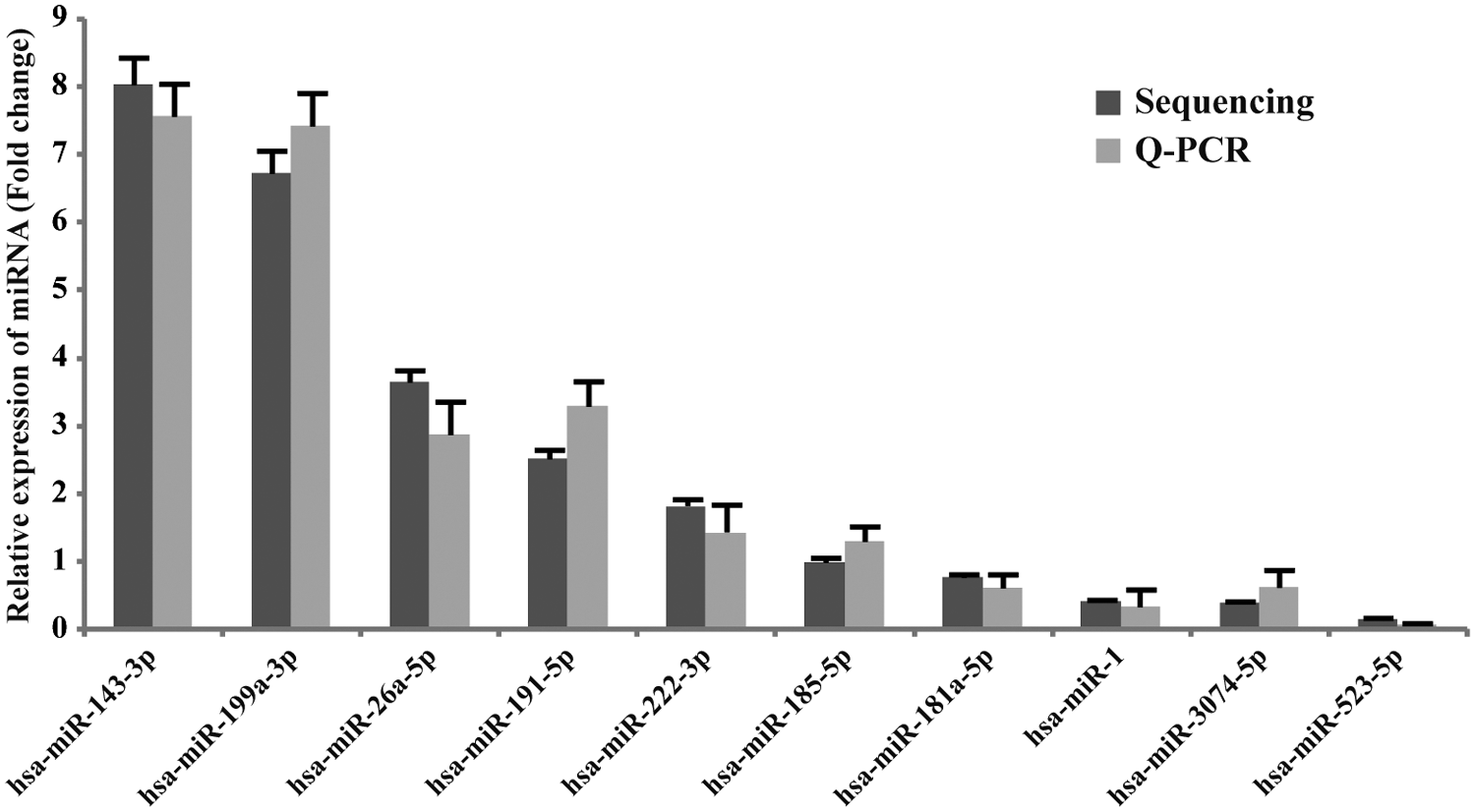
Confirmation of the 10 most abundantly expressed miRNAs in human ovaries by quantitative real-time PCR. Validation of the expression of 10 most abundantly expressed miRNAs by quantitative real-time PCR. The quantitative real-time PCR data with bars represent mean ± SD from three independent experiments. The sequencing data with bars represent the 95% CI. CI, confidence interval; miRNAs, microRNAs; PCR, polymerase chain reaction; Q-PCR, quantitative real-time PCR.
RPM, reads per million.
Recently, several reports suggested that miRNAs exhibit post-transcriptional enzymatic modification, such as 5′ or 3′ end trimming or additions of nucleotides and nucleotide changes of the mature miRNA sequence without a template, and these miRNA modifications may relate with miRNA stability or strengthen miRNA-mRNA interaction and even be involved in biological regulatory processes (Li et al., 2005; Azuma-Mukai et al., 2008; Morin et al., 2008; Ebhardt et al., 2009; Lu et al., 2009; Fernandez-Valverde et al., 2010). Therefore, we also analyzed the expression of miRNA isoform (including miRNA modification and editing) in human ovaries (Supplementary Tables S3 and S4). The miRNAs in the let-7 family showed the most abundant expression of miRNA modification and editing, which was in accordance with our previous reports (Tong et al., 2014; Xu et al., 2015), suggesting that the diversification of miRNA isoforms in let-7 family members in human ovaries might be involved in the processes of folliculogenesis and oogenesis (Miles et al., 2012).
Prediction of miRNA-targeted genes and identification of miRNA-regulated biological processes
To better understand the role played by the most abundantly known miRNAs in human ovaries, we identified the targeted genes, signaling pathways, and biological processes that could be targeted by these miRNAs. The putative target genes of miRNAs were predicted using miRanda, Targetscan, and MicroCosm, and these predicted miRNA targets were performed for GO and KEGG pathway analysis to enrich the involved signaling pathways and biological processes. After GO analysis, we found that the putative target genes of known miRNAs appeared to be involved in a broad range of processes, such as folliculogenesis-related processes (e.g., GO:0001547, antral ovarian follicle growth, and GO:0001543, ovarian follicle rupture), ovarian function-related processes (e.g., GO:0009566, fertilization, and GO:0007156, homophilic cell adhesion), and several key cellular signaling pathways (e.g., GO:0042990, regulation of transcription factor import into nucleus, GO:0045742, positive regulation of epidermal growth factor receptor signaling pathway, and GO:0008286, insulin receptor signaling pathway) (Table 3). Moreover, the known miRNA-related regulating pathways were enriched by KEGG pathway analysis (Table 4). Many ovarian function and oncogenesis-related signaling pathways were found to be involved, including the ErbB signaling pathway and Toll-like receptor signaling pathway.
GO, gene ontology.
Identification of novel miRNAs and prediction of their targeted genes and pathways
Recently, the NGS techniques have greatly revolutionized the detection of novel small RNAs with high levels of sensitivity and accuracy. Thus, the unclassified reads from NGS data were analyzed by Mireap and miRDeep to identify novel miRNAs in human ovaries. In this study, Mireap and miRDeep predicted the novel miRNAs based on default parameters with the read count more than 45, which were defined as novel miRNAs in human ovaries. Therefore, 21 novel miRNAs were identified in human ovaries, and the top 10 most abundant novel miRNAs are listed in Table 5. To confirm these 10 novel miRNAs obtained from NGS data, we used quantitative real-time PCR to validate them (Fig. 2). We further predicted the targeted genes of these novel miRNAs by miRanda, and these predicted targets were also performed for GO and KEGG pathway analysis. GO annotation for the predicted genes of novel miRNAs revealed that they may be involved in a broad range of biological processes related with the ovarian function, such as GO:0043409, negative regulation of MAPKKK cascade, and GO:0051897, positive regulation of protein kinase B signaling cascade (Supplementary Table S5). Similarly, several key pathways were also enriched according to KEGG pathway analysis, including the MAPK signaling pathway (Supplementary Table S6).
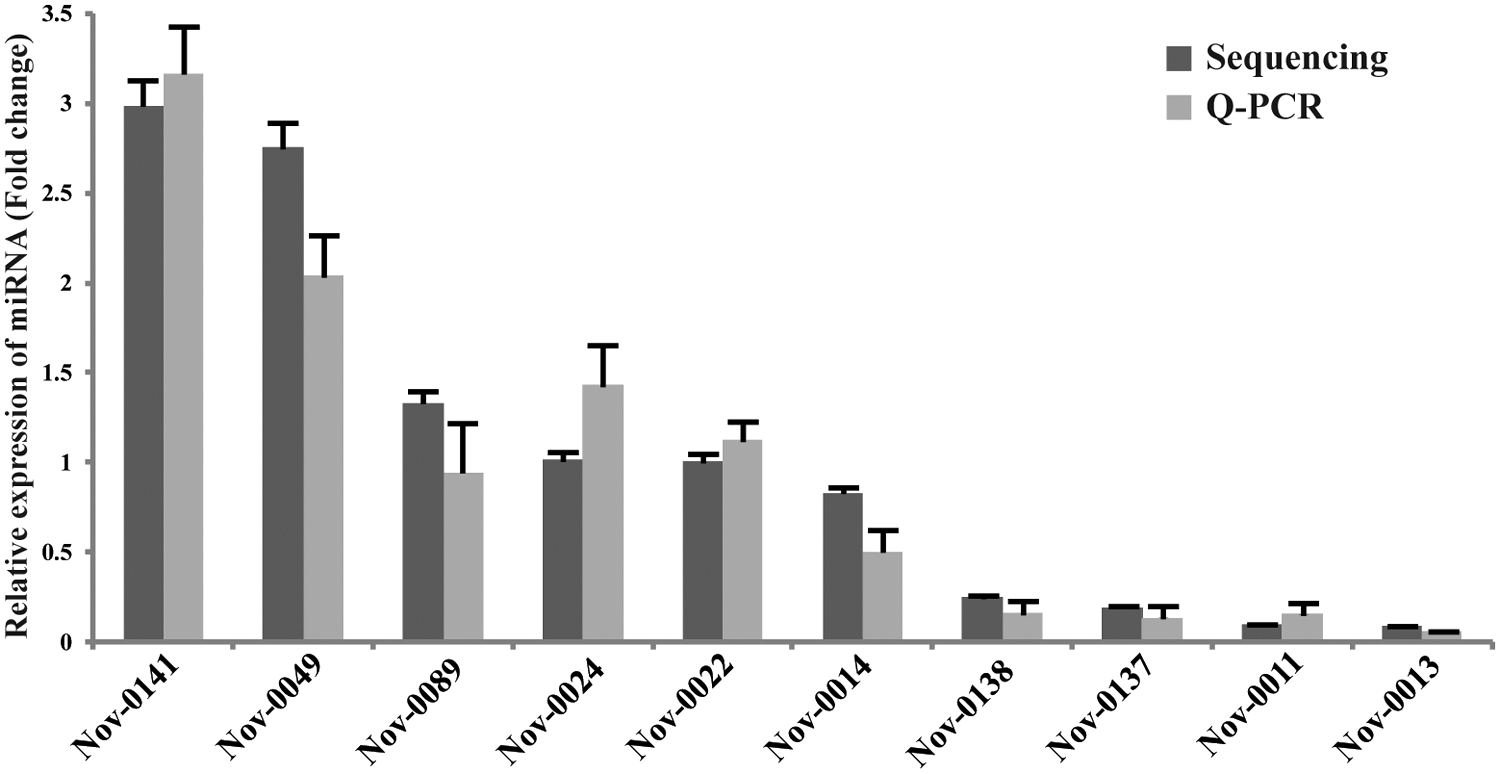
Validation of the expression of 10 most abundantly expressed novel miRNAs in human ovaries by quantitative real-time PCR. In human ovaries, the 10 expression levels of novel miRNAs were validated by quantitative real-time PCR. The real-time PCR data with bars represent mean ± SD from three independent experiments. The sequencing data with bars represent the 95% CI.
Discussion
Development of the mammal ovary is the result of a series of complex biological processes, such as oocyte growth, development, and differentiation of ovarian somatic cells, and interaction of oocytes and follicular cells (Carletti and Christenson, 2009; Hawkins and Matzuk, 2010; Hsueh et al., 2015; Li et al., 2015). Among the small noncoding RNAs associated with various ovary-related biological processes, miRNAs are best characterized as playing a key role during ovarian growth and function (Ahn et al., 2010; Lei et al., 2010; Li et al., 2015). In this study, for the first time, we analyzed the miRNA expression profile of human ovaries by NGS technology. Constructing expression profiles of small RNAs in human ovaries facilitates the understanding of their roles in the regulation of development and differentiation of ovaries.
Several known ovarian regulators or their receptors were reported to be targeted by miRNAs (Zhang et al., 2014; Xu et al., 2015). The let-7 miRNA family is expressed in the human, mouse, and cow ovaries and might be involved in regulation of oocyte maturation and oocyte–granulosa cell (GC) interaction in ovaries (Miles et al., 2012; Tong et al., 2014; Wagner et al., 2014; Xu et al., 2015), which were in accord with the has-let-7a-5p, has-let-7c-5p, and has-let-7e-5p and were the most abundant miRNAs expressed in human ovaries in this study. In previous reports, miR-199a and miR-140 were identified as the most downmodulated miRNAs in human ovarian cancer (Liu et al., 2013; Lan et al., 2015; Lian et al., 2015). In our sequencing data, miR-199a, miR-199b, and miR-140 were expressed in high levels in human ovary, suggesting that the maintenance of expression level of miRNAs in ovaries could be associated with the cancer formation or development. Ovarian GCs, surrounding the oocytes, are responsible for the development, maturation, and release of mature egg for fertilization and also responsible for synthesizing and secreting hormones (Hawkins and Matzuk, 2010; Tong et al., 2014; Zhang et al., 2014; Xu et al., 2015).
Recently, miRNAs have received widespread attention in ovarian GCs during folliculogenesis. For example, miR-320 was mainly expressed in GCs and oocytes of mouse ovarian follicles during folliculogenesis in a time-dependent manner (Feng et al., 2015). MiR-320 inhibited proliferation of GCs by targeting E2F1 and SF-1 (Yin et al., 2014). Moreover, miR-383 could promote the expression of miR-320 and enhance miR-320-mediated suppression of GC proliferation (Yin et al., 2014). In our study, the expression of miR-320a was also identified as one of the most abundant miRNAs in human ovaries. This suggested that the miRNAs, regulating the biological processes in GCs, take up a large slice of the miRNA expression profile of human ovary. Taken together, miRNAs and their function roles were generally involved in the physiological processes of ovaries.
In ovary life cycle, folliculogenesis is the most important process, including assembly of primordial follicles, follicle development, follicle rupture, and ovulation (Hsueh et al., 2015; Li et al., 2015). In this study, several folliculogenesis-related biological processes were identified for the ovary miRNA-targeted genes, such as antral ovarian follicle growth (GO:0001547), ovarian follicle rupture (GO:0001543), and fertilization (GO:0009566). Ovarian follicles are the basic units of female reproductive biology (Zhang et al., 2014; Hsueh et al., 2015). The formation, development, and maturation of follicles were also reported to be regulated by miRNAs (Lei et al., 2010; Zhang et al., 2014; Hsueh et al., 2015; Li et al., 2015). In mice, our previous report demonstrated that miR-376a regulates primordial follicle assembly by modulating the expression of Pcna (Zhang et al., 2014). When the primordial follicle pool is established, the size of this initial pool, in part, determines the reproductive life span of the female. These primordial follicles eventually develop into primary, secondary, and tertiary vesicular follicles (Zhang et al., 2014; Hsueh et al., 2015). During these processes of follicle development, miRNAs were reported to be involved in GCs and oocytes (Carletti and Christenson, 2009; Lei et al., 2010; Liu et al., 2010). For example, miR-181a could regulate GC proliferation and ovarian follicle development in mice (Zhang et al., 2013). Moreover, miR-224 is involved in the regulation of cumulus expansion and may affect ovulation and subsequent embryo development by targeting Ptx3 (Yin et al., 2014). Overall, folliculogenesis in human ovaries was regulated by miRNAs, and this study could provide a useful resource for further research of folliculogenesis in ovaries.
The ERK/MAPK pathway is involved in the regulation of various biological processes, including proliferation, differentiation, and cell cycle progression through several phosphorylation cascades (Mittal et al., 2009; Yamashita et al., 2013). In this study, the ERK/MAPK pathway was targeted by both the most abundantly known miRNAs and novel miRNAs in human ovaries, for example, GO:0045742, positive regulation of the epidermal growth factor receptor signaling pathway, and GO:0043409, negative regulation of MAPKKK cascade. Similarly, previous reports have demonstrated that the miRNAs participated in ovarian function through regulating the ERK/MAPK pathway. For example, overexpression of miR-15 reduced accumulation of proliferation- and apoptosis-related proteins, MAPK/ERK1, 2 and caspase 3, in GCs (Sirotkin et al., 2014). Moreover, hCG-mediated miR-122 expression is mediated by the activation of ERK signaling pathways (Menon et al., 2013).Our previous report also demonstrated that miR-485-5p could downregulate the expression of MAPK3 in PCOS cumulus GCs (Xu et al., 2015). Taken together, ovarian miRNAs regulate ovary-related processes, for example, proliferation of GCs, through the ERK/MAPK pathway.
PCOS, one of the most common endocrine disorders, is a multifactorial and heterogeneous syndrome with a constellation of symptoms and signs in human ovaries, such as arrest of follicle growth, decreased GC proliferation, and ovulatory dysfunction (Fux Otta et al., 2013). Although the underlying cellular mechanisms leading to PCOS remain unclear, PCOS is widely reported to be associated with increased risk of metabolic disorders, such as insulin resistance, diabetes, and obesity (Fux Otta et al., 2013; Sorensen et al., 2014). Our previous report has demonstrated that miRNAs and their targeted pathways play important roles in the etiology and pathophysiology of PCOS (Xu et al., 2015). In this study, the insulin-related biological process (insulin receptor signaling pathway, GO:0008286) was also identified as the targeted pathway for the miRNAs in human ovaries. Compared with controls, miR-132 and miR-320 were identified that have decreased expression in the follicular fluid of women with PCOS (Onalan et al., 2005). Moreover, miRNA-21, miRNA-27b, miRNA-103, and miRNA-155 were differentially expressed in PCOS serum (Murri et al., 2013). These results suggested that miRNA in ovaries participates in regulation of not only physiological processes but also reproductive disorders, for example, PCOS.
In summary, for the first time, we have identified the known and novel expressed miRNA profiles of human ovarian tissues directly by high-throughput Solexa sequencing. Moreover, we validated the most abundantly expressed miRNAs and the predicted novel miRNAs in human cumulus GCs by quantitative real-time PCR. The GO term annotation and KEGG pathway analysis for the predicted miRNA targets further indicate that these miRNAs (most abundant) are involved in various biological processes, such as antral ovarian follicle growth, ovarian follicle rupture, and fertilization. Moreover, these miRNAs are also indentified as regulators in etiology and pathophysiology of PCOS. These results suggested that miRNAs play important roles in the development and physiological function of human ovary. Our work supports and further extends the knowledge of a regulatory role of miRNAs and their targeted processes in human ovaries and may also offer new insights for the treatment of reproductive and ovarian disorders.
Footnotes
Acknowledgments
This research was supported by the Natural Science Foundation of Anhui Province of China (1308085QH131, 1408085MH150), the National Natural Science Foundation of China (81370757), and Natural Science Research Project of the Anhui Provincial Education Department (KJ2013Z134).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.