
Abstract
Estrogen signals have been suggested to modulate the progression and metastasis of nonsmall cell lung cancer (NSCLC), which is one of the leading causes of cancer deaths worldwide. While there are limited data concerning the roles and effects of G-protein-coupled estrogen receptor (GPER) on the progression of NSCLC, our present study reveals that the expression of GPER in NSCLC cells is obviously greater than that in lung fibroblast cell line MRC-5. Activation of GPER via its specific agonist G-1 decreases the in vitro motility of A549 and H358 cells and the expression of matrix metalloproteinase 2 (MMP-2) and MMP-9. Further, G-1 treatment can rapidly decrease the phosphorylation, nuclear translocation, and promoter activities of NF-κB in NSCLC cells. BAY 11-7082, the inhibitor of NF-κB, also inhibits the expression of MMP-2/9, while overexpression of p65 significantly attenuates G-1-induced downregulation of MMP-2/9. It suggests that inhibition of NF-κB mediates G-1-induced MMP-2/9 downregulation. G-1 treatment significantly down regulates the phosphorylation of IκB kinase β (IKK-β) and IκBα, while not IKK-α, in both 549 and H358 cells. ACHP, the specific inhibitor of IKK-β, can reinforce G-1-induced MMP-2/9 downregulation and invasion suppression of A549 cells. Collectively, our results suggest that activation of GPER can inhibit the migration of human NSCLC cells via suppression of IKK-β/NF-κB signals. These findings will help to better understand the roles and mechanisms of GPER as a potential therapy target for NSCLC patients.
Introduction
L
Recently, several epidemiologic and clinical studies have provided evidence of a role for estrogen in the genesis and progression of NSCLC (Stabile et al., 2002; Hershberger et al., 2005). It is reported that women have a higher risk of lung adenocarcinoma, a type of NSCLC, compared to men, independent of smoking status (Ramchandran and Patel, 2009; Kiyohara and Ohno, 2010). Women taking antiestrogens have a significant lower risk of developing lung cancer (Bouchardy et al., 2011). Estrogen can promote lung cancer growth and metastasis both in vitro and in vivo (Hershberger et al., 2005; Schwartz et al., 2005). All these data suggested that estrogen signals can modulate the progression and metastasis of NSCLC.
The G-protein-coupled estrogen receptor (GPER), formerly known as GPR30, is first identified as a G-protein-coupled receptor (GPCR) involved in membrane-mediated E2 signaling (Prossnitz et al., 2008; Prossnitz and Maggiolini, 2009). GPER is reported to bind E2 with high affinity (Kd = 3 ∼ 7 nM) and to activate multiple intracellular signal transduction pathways, including adenylyl cyclase, epidermal growth factor receptors (EGFRs), mitogen-activated protein kinase (MAPK), and phosphoinositide 3-kinase (PI3K) signaling pathways (Ge et al., 2012; Luo et al., 2012). To date, there are only few reports about GPER and motility of cancer cells providing confusing and controversial results. Activation of GPER can suppress the migration of ovarian cancer cells (Henic et al., 2009; Ignatov et al., 2013), whereas in other studies, activation of GPER is able to promote the metastasis of ER-negative breast (Pandey et al., 2009; Yu et al., 2014), ovarian (Yan et al., 2013), and endometrial (He et al., 2012) cancer cells. Therefore, the roles and mechanisms of GPER in NSCLC cell migration and invasion need further studies.
Recently, studies reported that the expression of GPER is higher in lung tumors compared to normal tissue (Jala et al., 2012). Little is known regarding the function of elevated GPER in lung cancer. Therefore, the present study is designed to investigate effects of GPER activation on migration and invasion of NSCLC. Furthermore, the downstream signal pathways of GPER in NSCLC cell lines are further investigated.
Materials and Methods
Cell lines and regents
Human NSCLC cell lines A549, H358, H1793, H1395, CCL-185, lung fibroblast cell line MRC-5, and human breast cancer cell line MCF-7 were purchased from the American Type Culture Collection (ATCC) and used within 10 passages from the time of purchase from ATCC. All chemicals and inhibitors were purchased from Sigma Chemical Co., unless otherwise noted. Monoclonal antibodies were purchased from Cell Signaling Technology, Inc. and the horseradish peroxidase-conjugated secondary antibody from Santa Cruz Biotechnology. All compounds were solubilized in dimethyl sulfoxide (DMSO) with the concentrations of DMSO in the assay less than 0.5% (v/v). A medium containing 0.5% DMSO was used as the control.
Cell culture and transfection
Human cancer cells were cultured in DMEM or RPMI 1640 with 10% fetal bovine serum, 100 U/mL penicillin, and 100 g/mL streptomycin at 37°C in a humidified 5% CO2 atmosphere. For experiments, cells were plated in a phenol red-free medium for at least 24 h before treatment. For transfection, cells were seeded into six-well plates in 2 mL of growth medium and transfected with pcDNA3.1 (vector control) or pcDNA3.1/p65 by use of Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. The transfection efficiency was analyzed by Western blot analysis after transfection for 24 h.
Cell proliferation assay
Cell viability was measured using the 3-(4, 5-Dimethylthiazol-2-yl)-2, 5- Diphenyltetrazolium Bromide (MTT) assay according to the previous study (Ge et al., 2014). Briefly, cells were seeded in 96-well plates with 5 × 103 cells per well in 100 μL of cell culture medium and incubated with various concentrations of G-1 for the indicated times. Then, the medium was removed and the precipitated formazan was dissolved in 100 μL of DMSO. After shaking the incubation for 10 min, the absorbance at 570 nm was detected using a uQuant Universal Microplate Spectrophotometer (Bio Tek Instruments). Cell viability was expressed as a percentage of the absorbance value of control cultures. The experiments were repeated six times. The 50% inhibitory concentration (IC50) was calculated using GraphPad Prism software (GraphPad Software, Inc.).
Wound healing assay and in vitro invasion assays
For the in vitro wound healing assay, confluent monolayers of A549 and H358 cells were scratched with three separate wounds by use of a sterile 200 μL pipette tip. The migration distance of the cells into the scratched area was measured in five randomly chosen fields. Pictures were taken just above and just below each line to ensure that the line just appears in each picture. For in vitro invasion assay, polycarbonate filters (8 μm pore size; Corning) were precoated with Matrigel Matrix (BD Biosciences). Cells (1 × 105) treated with or without G-1 in 200 μL medium (containing 0.1% FBS) were seeded in the upper chamber. Then, 600 μL medium with 10% FBS were added to the lower chamber and served as a chemotactic agent. After incubation for the indicated times, cells adhering to the underside of the filter were stained with hematoxylin and counted under an upright microscope (five fields per chamber). The cell invasion index of each group was calculated as the percentage of control group.
Quantitative real-time PCR analysis
Total RNA samples were extracted from cells using the TRIzol reagent (Invitrogen). The RNA was converted into cDNAs using the RevertAid First Strand cDNA Synthesis Kit (Fermentas, K1621). Quantitative real-time PCR (qRT-PCR) was run on an iCycler (Bio-Rad) using validated primers and SYBR Premix Ex Taq II (Takara) for detection. The cycle number, when the fluorescence first reached a preset threshold (Ct), was used to quantify the initial concentration of individual templates for expression of mRNA of genes of interest. Transcripts of the housekeeping gene GAPDH in the same incubations were used for internal normalization. Primer sequences were as follows: VEGF, 5′-TCG AGT ACA TCT TCA AGC CAT CCT G-3′ and 5′-TCC TAT GTG CTG GCC TTG GTG AG-3′; GAPDH, 5′-TGA ACG GGA AGC TCA CTG G-3′ and 5′-TCC ACC ACC CTG TTG CTG T-3′. All experiments were performed three times independently and the average was used for comparison.
Western blot analysis
Cells treated with or without G-1 were detached by trypsin, washed three times with PBS, and treated with a lysis buffer (25 mM Tris–HCl pH 7.4, 1% Triton X-100, 150 mM NaCl, 5% EDTA, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride, and 10 mg of aprotinin and leupeptin). Aliquots of protein were separated on 10% sodium dodecyl sulfate (SDS)–polyacrylamide gels and electrophoretically transferred to nitrocellulose membranes. Following blocking with 5% nonfat milk at room temperature for 2 h, membranes were incubated with primary antibodies at 1:1000 dilution overnight at 4°C, then incubated with a horseradish peroxidase-conjugated secondary antibody at 1:1000 dilution for 2 h at room temperature, and detected with the Western Lightning Chemiluminescent detection reagent (Perkin-Elmer Life Sciences). GADPH was used as an internal control for protein loading and analysis.
NF-κB reporter assay
A549 cells were transiently cotransfected with pNF-κB-luc (2 μg) and pRL-TK (0.5 μg). After 24 h, these cells were treated with G-1 for the indicated times. Transcriptional activity was determined by the dual-luciferase reporter assay system. Results were calculated as the ratio between the activity of pNF-κB-luc and pRL-TK.
Statistical analysis
Statistical analysis was performed using the IBM SPSS, version 19 (SPSS, Inc.). Student's t-test was used to evaluate the statistical significance of the in vitro studies. p values <0.05 were considered statistically significant.
Results
The expression of GPER in NSCLC cells
To investigate the effect of GPER on the progression of NSCLC, its protein expression and mRNA expression were measured in various NSCLC cell lines and compared with the positive control (MCF-7 cells) and lung fibroblast cell line MRC-5. Our results showed that the protein levels of GPER in NSCLC cell lines were obviously greater than that in lung fibroblast cell line MRC-5, and comparable with or slightly less than that in MCF-7 cells (Fig. 1A). This was confirmed by the results of qRT-PCR; the mRNA levels of GPER in NSCLC cells were greater than that in MRC-5 cells. A549 and H358 cell lines were chosen for further study due to the relative high levels of GPER. These data suggested that GPER is also highly expressed in human NSCLC cells.
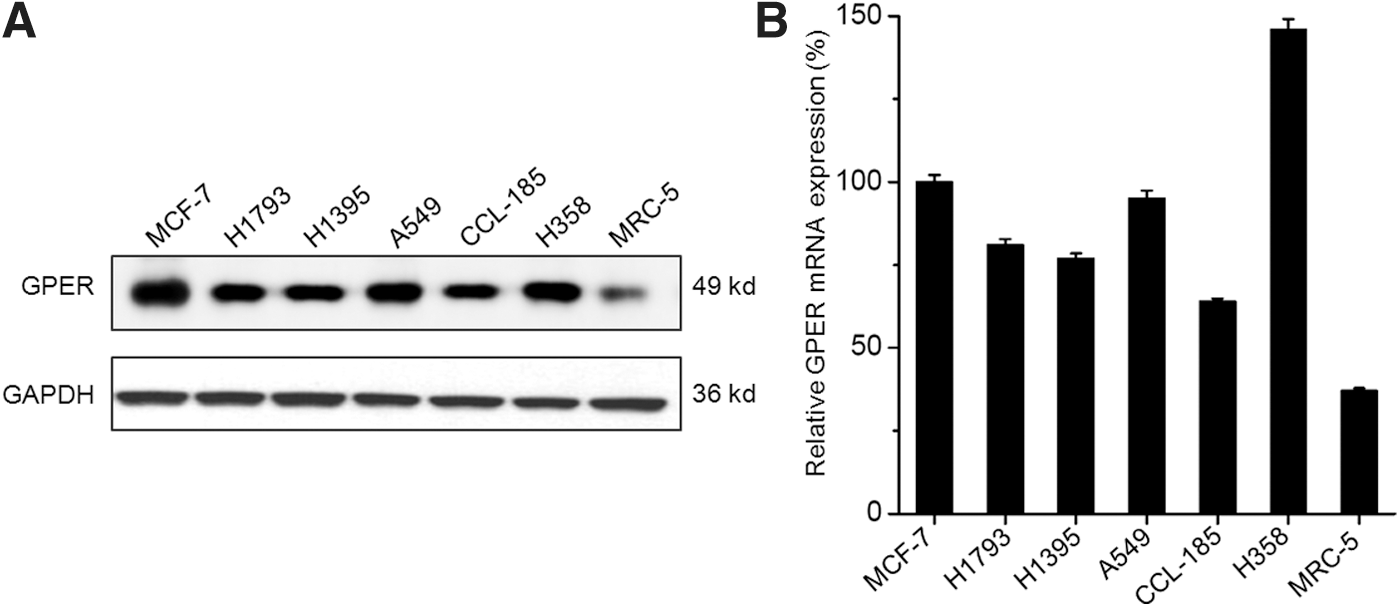
The expression of GPER in NSCLC cell lines.
Activation of GPER can suppress the in vitro motility of NSCLC cells
To investigate the activation of GPER on the progression of NSCLC, we further tested the dose–response of GPER agonist on the proliferation of A549 and H358 cells. The results showed that G-1 can inhibit the proliferation of both A549 and H358 cells via a concentration-dependent manner (Fig. 2A). The IC50 of G-1 in A549 and H358 cells at 24 h was 4.1 and 2.5 μM, respectively. According to the previous studies (Dennis et al., 2011; Vaucher et al., 2014) and cell viability assay, the concentration of 200 nM was chosen for further cell motility studies because it has no significant effect on cell proliferation.
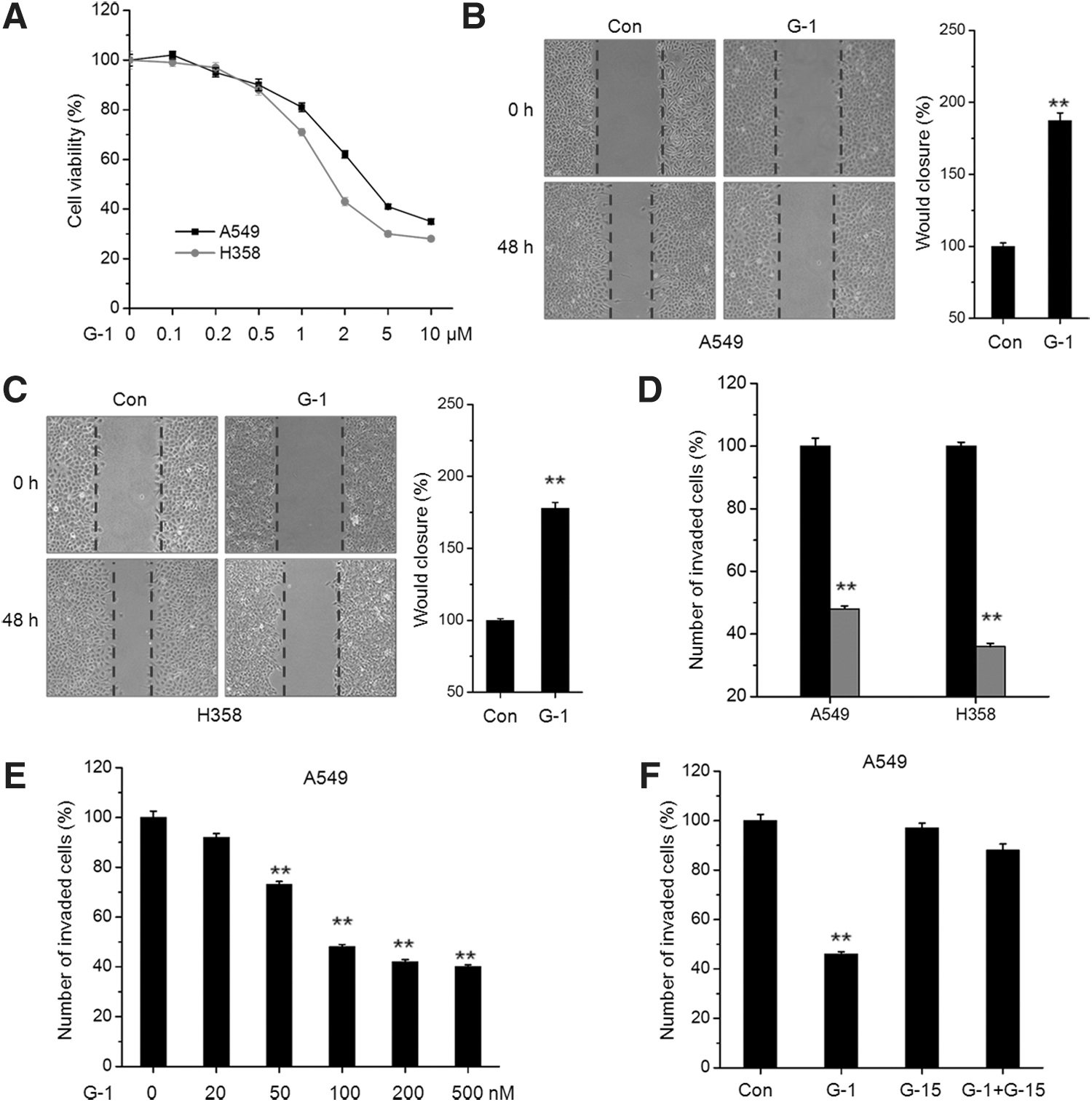
Activation of GPER inhibits the in vitro motility of NSCLC cells.
Then, we further investigated the effects of G-1 on the in vitro motility of NSCLC cells by use of wound healing and transwell invasion assays. The results revealed that G-1 at 200 nM significantly decreased wound closure compared to the control group for both A549 (Fig. 2B) and H358 (Fig. 2C) cells. It was confirmed by the results of invasion assay, which showed that G-1 at 200 nM can significantly (p < 0.01) inhibit the in vitro invasion of A549 and H358 cells (Fig. 2D). Furthermore, G-1 treatment can increase the in vitro invasion of A549 cells via a dose-dependent manner (Fig. 2E), while the inhibition effects of G-1 on cell invasion can be attenuated by a GPER-specific inhibitor G-15 (Fig. 2F). Collectively, our data revealed that activation of GPER can inhibit the in vitro motility of NSCLC cells.
G-1 inhibits the expression of migration-related proteins
To investigate the mechanisms responsible for the suppression effects of G-1 on in vitro cell motility, we further tested its effect on the expression of cell migration-related proteins, including matrix metalloproteinase 2 (MMP-2), MMP-9, Vim, and FN (Caccavari et al., 2010; Bauvois, 2011). Our results showed that G-1 treatment significantly inhibited the expression of MMP-9 and MMP-2 in both A549 (Fig. 3A) and H358 (Fig. 3B) cells, whereas had limited effects on the expression of Vim and FN. Furthermore, G-1 treatment can down regulate the mRNA expression of MMP-2 and MMP-9 via a time-dependent manner in both A549 and H358 cells (Fig. 3C). These data suggested that G-1 can inhibit the expression of MMP-2 and MMP-9 and then suppress the in vitro motility of NSCLC cells.
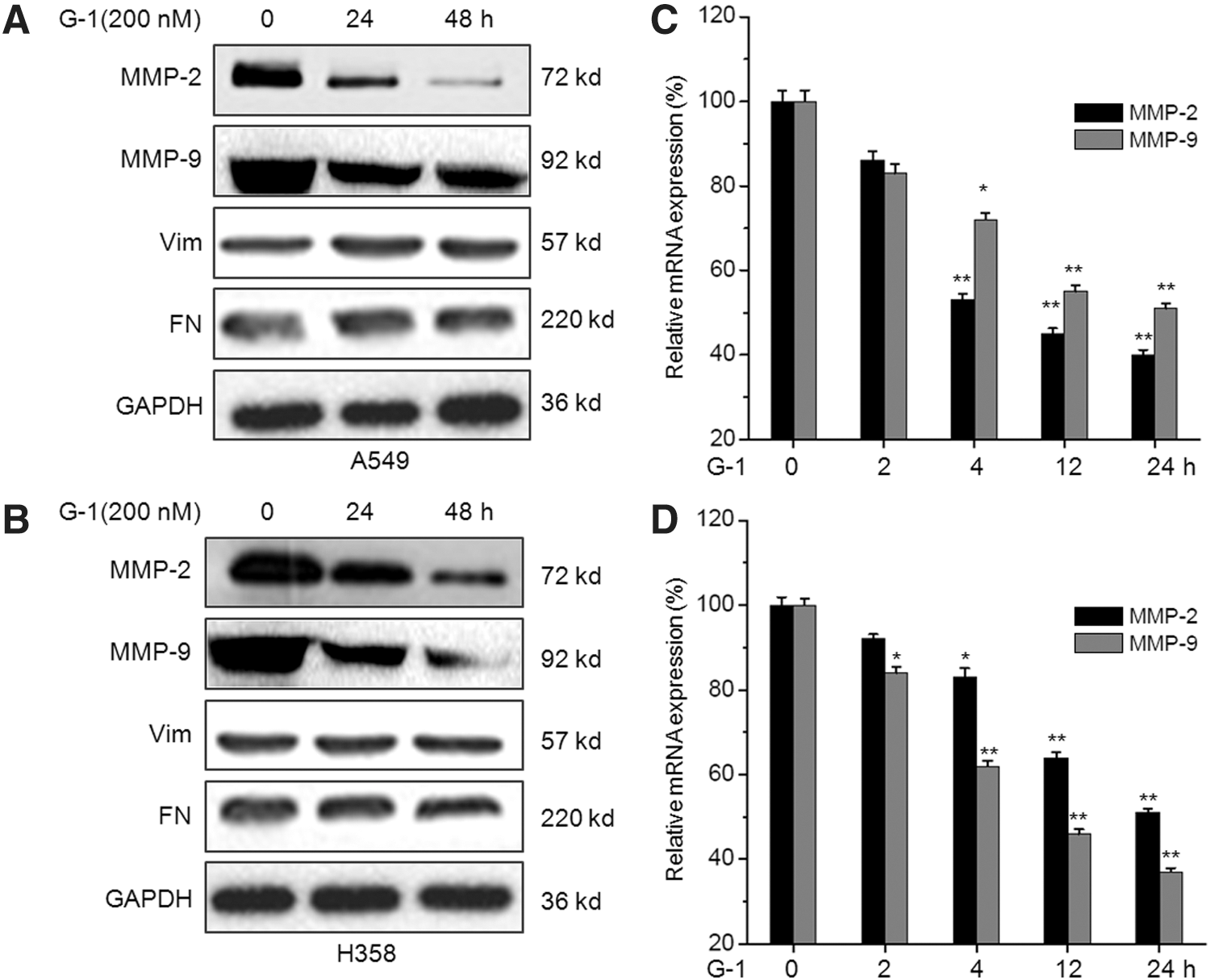
Activation of GPER via G-1 inhibits the expression of migration-related proteins in NSCLC cells. A549
G-1 can suppress the activities of NF-κB in NSCLC cells
Numerous studies indicated that NF-κB is a key transcription factor for cell migration and MMP expression (Bond et al., 2001; Min et al., 2008). Therefore, we investigated the effect of G-1 on the phosphorylation and subcellular location of NF-κB in NSCLC cells. The results showed that G-1 treatment can rapidly decrease the phosphorylation of p65 in both A549 and H358 cells (Fig. 4A). Since NF-κB activation requires the translocation of its p65 subunit to the nucleus, we measured the levels of p65 in the cytoplasm and in the nucleus in G-1-treated cells. The results indicated that G-1 treatment obviously decreased the levels of p65 in the nucleus of A549 cells (Fig. 4B). Furthermore, the suppression of NF-κB by G-1 was further verified by time dependently increasing the NF-κB promoter activities in both A549 (Fig. 4C) and H358 (Fig. 4D) cells. Collectively, these data suggested that activation of GPER via G-1 can suppress the activation and nuclear translocation of NF-κB in NSCLC cells.
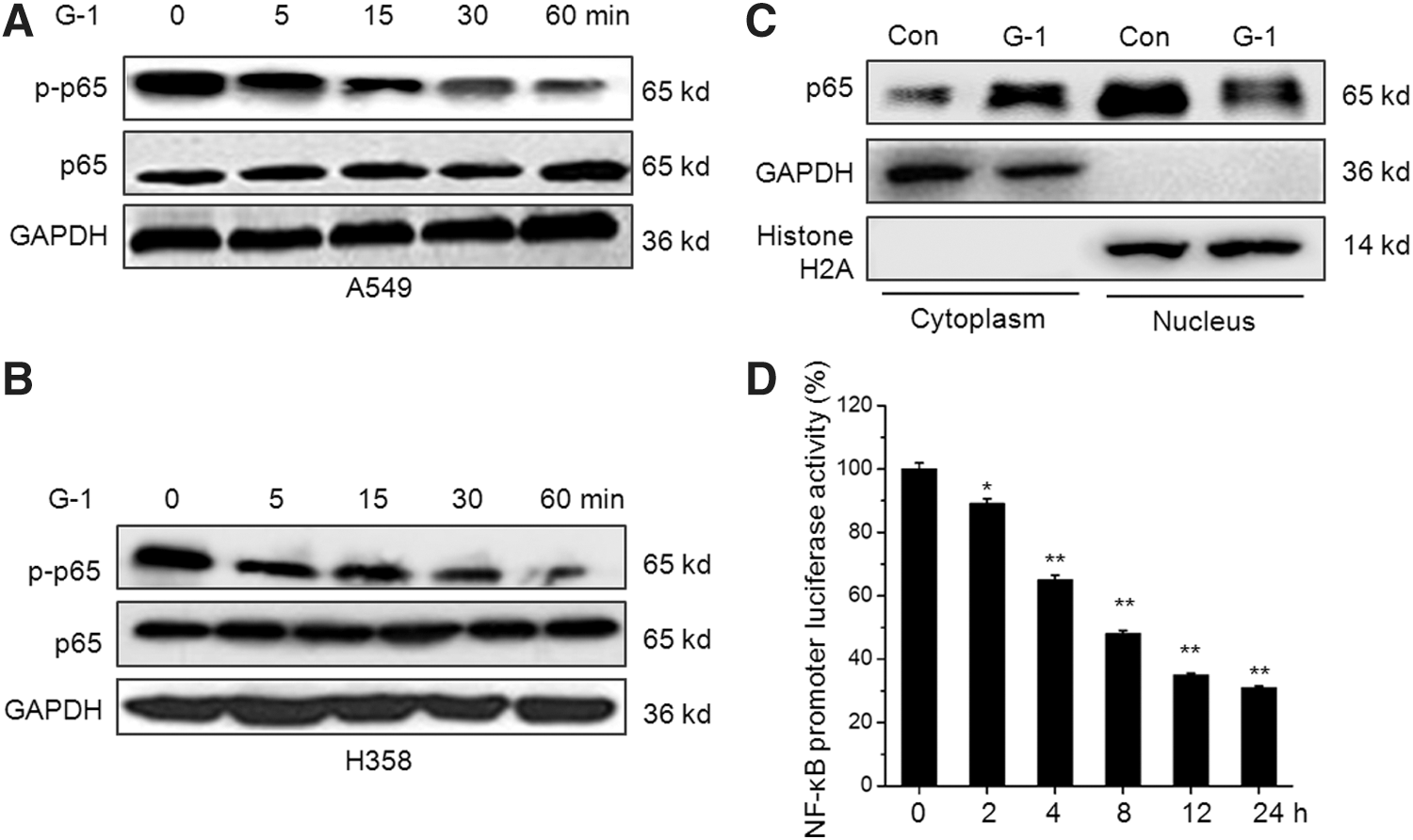
G-1 suppresses the activities of NF-κB in NSCLC cells. A549
The inhibition of NF-κB mediates G-1-induced MMP-2/9 downregulation
To verify the roles of NF-κB in G-1-induced MMP-2/9 suppression of NSCLC cells, we treated both A549 and H358 cells with BAY 11-7082, the inhibitor of NF-κB, for 24 h. Then, the expression of MMP-2/9 was measured by Western blot analysis. The results suggested that BAY 11-7082 can also inhibit the expression of MMP-2/9 in both A549 and H358 cells (Fig. 5A). Furthermore, we transfected pcDNA3.1/p65 to overexpression of NF-κB in A549 and H358 cells, which was confirmed by the results of Western blot analysis (Fig. 5B). Our results showed that overexpression of p65 significantly attenuated G-1-induced downregulation of MMP-2/9 in both A549 and H358 cells (Fig. 5C). Collectively, our results revealed that NF-κB mediates the suppression effects of G-1 on MMP-2/9 expression of NSCLC cells.
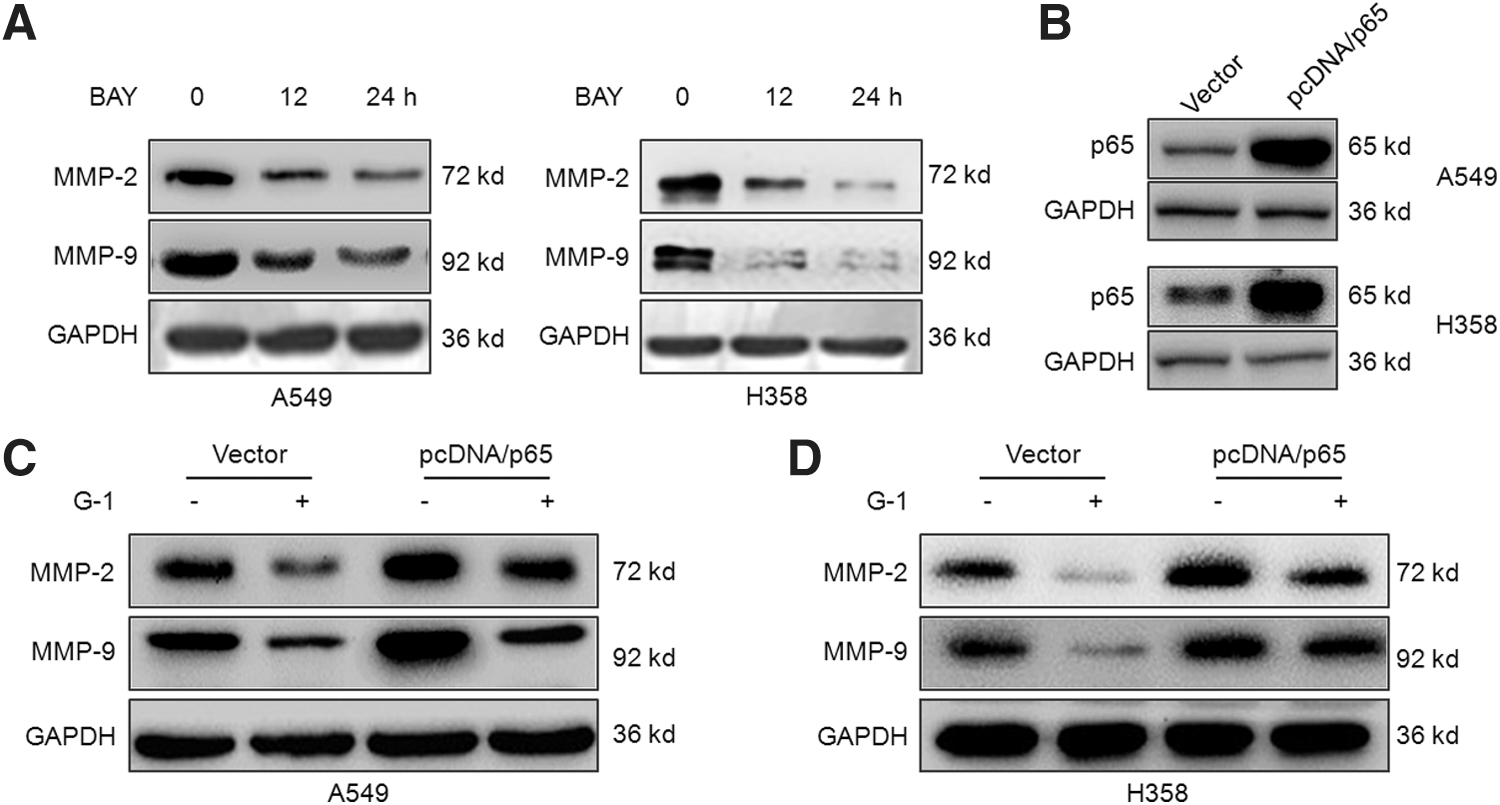
The inhibition of NF-κB mediates G-1-induced MMP-2/9 downregulation.
G-1 decreases the activity of NF-κB and MMP-2/9 expression via dephosphorylation of IKK-β
Previous studies indicate that IKK-α and IKK-β are required for the phosphorylation of IκBα and p65 in cancer cells (Woronicz et al., 1997; Nottingham et al., 2013). Then, we measured the effects of G-1 on the phosphorylation of IKK-α, IKK-β, and IκBα in both A549 and H358 cells. The results showed that 200 nM G-1 treatment significantly down regulated the phosphorylation of IKK-β and IκBα, while not IKK-α, in both 549 and H358 cells (Fig. 6A). Furthermore, ACHP, the specific inhibitor of IKK-β, can inhibit the MMP-2/9 and cooperatively reinforce G-1-induced MMP-2/9 downregulation in A549 cells (Fig. 6B). Similar results were also observed for the effects of G-1 on in vitro invasion of A549 cells (Fig. 6C). These data suggested that the inhibition of IKK-β is involved in G-1-induced MMP-2/9 and cell motility downregulation of NSCLC cells.
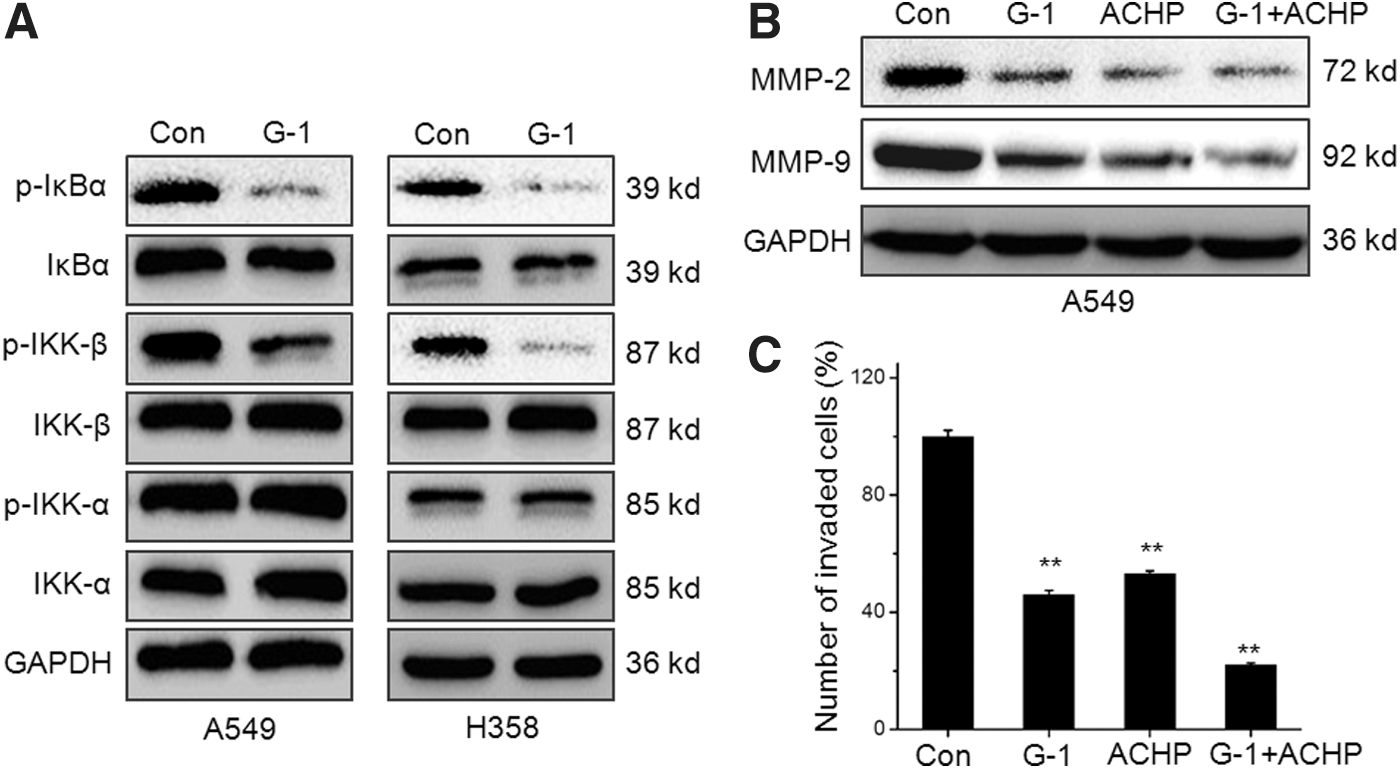
G-1 decreases the activity of NF-κB and MMP-2/9 expression via dephosphorylation of IKK-β.
Discussion
Although classical estrogen receptors can be detected in lung cancer cells and promote the development (Siegfried and Stabile, 2014), there are limited data concerning the expression and functions of GPER in the metastasis of NSCLC cells. Our present study indicates that the expression of GPER in NSCLC cells is greater than that in the lung fibroblast cell line MRC-5. Activation of GPER via its agonist G-1 can suppress the in vitro motility and expression of MMP-2/9 of NSCLC cells. Furthermore, G-1 treatment obviously inhibits the activation, nuclear translocation, and transcriptional activities of NF-κB, while overexpression of p65 attenuates G-1-induced downregulation of MMP-2/9. G-1 treatment also significantly suppresses the phosphorylation of IKK-β and IκBα, while not IKK-α. ACHP, the specific inhibitor of IKK-β, can reinforce G-1-induced MMP-2/9 downregulation and invasion suppression of A549 cells. Collectively, our results suggest that activation of GPER can inhibit the migration of human NSCLC cells via suppression of IKK-β/NF-κB signals.
Recently, more and more evidences supporting GPER, a newly discovered membrane-bound estrogen receptor and member of the GPCR family, as a tumor suppressor signal (Chan et al., 2010; Holm et al., 2011; Wang et al., 2013; Wei et al., 2014). Our results that activation of GPER inhibits the in vitro cell motility of NSCLC cell are similar with the results in ovarian cancer (Ignatov et al., 2013), while contradictory with other studies that activation of GPER is able to promote the metastasis of ER-negative breast (Pandey et al., 2009; Yu et al., 2014), ovarian (Yan et al., 2013), and endometrial (He et al., 2012) cancer cells. This might be because the stimulatory effects of GPER are stimulated with nonspecific agonists such as estrogen and tamoxifen rather than its specific agonist G-1. Considering that both ERα/β and estrogen-related receptor (ERR) are expressed, it appears that the major migration and invasion effects are exerted and promoted by ERα (Ye et al., 2010) or ERRα (Wu et al., 2015). Our data were supported by a recent study that activation of GPER via G-1 can suppress the progression of lung cancer cells via oxidative/antioxidative enzyme-mediated antiproliferative and proapoptotic effects (Kurt et al., 2016). A recent study revealed that GPER is overexpressed in lung tumors and lung adenocarcinoma cell lines relative to normal lung and immortalized normal lung cell lines (Jala et al., 2012). It might be that overexpression of GPER in lung cancers may reflect a defense mechanism to counteract excessive proliferation. The loss of GPER in breast (Chen et al., 2016) and endometrial (Gao et al., 2011) cancers is associated with poor prognosis. Considering that the effects of GPER on cell motility seem to be cell line dependent, further studies are needed to confirm the roles of GPER on metastasis of other cancers.
The inhibition effects of G-1 on cell migration were accompanied by downregulation of the migration-related factors MMP-2 and MMP-9. Several studies revealed expressions of MMP-2 and MMP-9 are strongly associated with tumor metastasis and negatively correlated with patient's survival (Bauvois, 2011). Therefore, inhibition of MMPs is suggested to be a promising therapeutic approach for lung cancer treatment (Pinto et al., 2003; Gueders et al., 2006). The expression and activation of MMP-2 and MMP-9 are complexly controlled by upstream signaling pathways, including MAPK, PI3K/Akt, and NF-κB (Kondapaka et al., 1997; Min et al., 2008). Our study revealed that G-1 treatment can significantly inhibit the activation, nuclear translocation, and transcriptional activities of NF-κB, while overexpression of p65 attenuates G-1-induced downregulation of MMP-2/9. NF-κB has been identified as an important regulator of metastasis of cancer cells (Huber et al., 2004; Maier et al., 2010). Furthermore, there exist multiple NF-κB consensus binding motifs in the promoter of MMP-2 and MMP-9 (Cheng et al., 2006). Therefore, our study revealed that activation of GPER via G-1 can inhibit the activities of NF-κB and then down regulate the expression of MMP-2/-9 to suppress the in vitro motility of NSCLC cells. The expression of MMP-2 and MMP-9 can also be controlled by several other transcription factors such as AP-1 and SP1 (Hwang and Park, 2010). Whether these factors are involved in G-1-induced downregulation of MMP-2 and MMP-9 needs further studies.
IKK-β is the major kinase responsible for phosphorylation of all the IκB subunits (Mercurio et al., 1997). Our study also reveals that IKK-β, while not IKK-α, is involved in G-1-induced downregulation of p65. The specific inhibitor of IKK-β can inhibit expression of MMP-2/-9 and cooperatively reinforce the suppression effect on NSCLC invasion. These are consistent with the previous studies showing that IKK-β plays an important role in the anticancer activities of many agents (Bernier et al., 2006; Wu et al., 2013). It has been suggested that a variety of upstream activating kinases participate in the signaling cascade leading to IKK-β activation (Woronicz et al., 1997). Further research work is necessary to investigate how activation of GPER via G-1 can inhibit the IKK-β activation.
In conclusion, GPER has been highly detected in NSCLC cells. Its activation by specific agonist can inhibit the in vitro motility of NSCLC cells via downregulation of MMP-2 and MMP-9. The inhibition of NF-κB via IKK-β mediates the suppression effects of G-1 on the invasion of NSCLC cells and expression of MMP-2 and MMP-9. Although the precise mechanism needs further studies, these findings will help to better understand the roles and mechanisms of GPER as a potential therapy target for NSCLC patients.
Footnotes
Acknowledgments
This study was partly supported by a grant (No. 81241071) from the National Nature Science Foundation of China (The research on mechanism of IκB regulated by NF-κB on reversing malignant phenotype of different lung carcinoma cells.) and a grant (No. 7112039) from the Beijing Natural Science Foundation. (The research on mechanism of IκB regulated by NF-κB on reversing malignant phenotype of human lung adenocarcinoma cells.)
Disclosure Statement
The authors declare no conflicts of interest.