
Abstract
Pathogenesis of Crohn's disease (CD) is thought to involve the chronic activation of T helper 1 (Th1)- and Th17-mediated inflammation, such as the production of interferon-gamma (IFN-γ) and interleukin 17 (IL-17). However, studies have also shown that although IFN-γ is required, IFN-γ-producing or T-bet-expressing Th1 cells are dispensable. We therefore examined T-bet-expressing B cells as another source of IFN-γ that potentially supported intestinal inflammation in CD patients. We found that the frequencies of T-bet-expressing B cells were significantly upregulated and abundantly present in the gut of active, but not quiescent, CD patients. The frequencies of T-bet-expressing B cells were also directly correlated with CD disease activity. These T-bet+ B cells were almost exclusively IgG expressing and produced significantly higher amounts of IFN-γ, IL-6, and IL-12 than IgA- and IgM-expressing T-bet− B cells. These B cells also supported IFN-γ production of CD4+ T cells. T-bet expression was induced in vitro in peripheral blood B cells through the stimulation of B-cell receptor (BCR), Toll-like receptor 7 (TLR7), and IFN-γ, which resembled gut T-bet+ B cells in terms of elevated IFN-γ. We found that these stimulated B cells, but not unstimulated B cells, supported the IFN-γ and IL-12 production from autologous CD4+ T cells. In addition, in patients with higher gut T-bet+ B-cell percentage, a higher frequency of gut-infiltrating IFN-γ+ and IL-12+ T cells was also observed. Together, our results suggested that T-bet-expressing B cells could contribute to the intestinal Th1 inflammation in CD patients.
Introduction
C
In addition to mediating Th1 differentiation and responses, T-bet is also expressed by other cell types, including CD8+ T cells, dendritic cells, natural killer (NK) cells, and B cells (Lazarevic et al., 2013). Here, we are particularly interested in the T-bet-expressing B cells because B cell and its effector form, antibody-secreting plasmablasts, make up the majority of lymphocytes in the intestinal lamina propria and the Peyer's patches(Cerutti et al., 2011; Mowat and Agace, 2014). At steady state, the vast majority of intestinal B cells express IgA, with a small minority expressing IgM and very few expressing IgG (Cerutti et al., 2011; Mowat and Agace, 2014). Binding of IgA on microbial organisms in the gut is thought to promote homeostasis with commensals without activating an inflammatory response. However, in inflammatory bowel diseases, including CD and ulcerative colitis, the frequencies of IgG-expressing B cells and the levels of secreted IgG are significantly elevated (Macpherson et al., 1996; Shulzhenko et al., 2011). Interestingly, T-bet expression in B cells promotes the class switching toward the IgG2a isotype and mediates the formation of IgG2a-expressing memory B cells (Peng et al., 2002; Wang et al., 2012). T-bet in B cells also upregulates IFN-γ, which, in turn, is an IgG1 and IgG2 class-switch factor (Kawano et al., 1994; Harris et al., 2005). These results suggest that T-bet-expressing B cells are present at the CD intestinal tract and can potentially act as a source of IFN-γ. Whether T-bet-expressing B cells can directly contribute to CD pathogenesis is unknown.
Therefore, in this study, we recruited active CD patients and examined the role of T-bet-expressing B cells.
Materials and Methods
Recruitment of participants
Thirty-three CD patients were recruited, including 20 active patients (Crohn's Disease Activity Index [CDAI] score ≥220 and CRP level ≥2 times upper normal levels) and 13 quiescent patients (defined as CDAI score <150). The presence of inflammation was confirmed by endoscopy, ultrasonography, and/or computed tomography scanning. All examinations were conducted and interpreted by experts in inflammatory bowel diseases according to previously established criteria (Van Assche et al., 2010). Additional inclusion criteria included ages between 18 and 65 years, blood leukocyte >4 × 109 cells/L, CD4+ T > 300 cells/mm3, platelet >150 × 109 cells/L, hemoglobin >10.5 g/L, and serum creatine within normal ranges. Twelve age- and sex-matched healthy controls were then recruited. Subjects with active infections, cardiovascular diseases, HBV infections, diabetes, concomitant therapies with immunosuppressants, and malignancies were excluded. The review board of the Jinling Hospital approved the use of peripheral blood and gut biopsy samples. Written informed consent was obtained from every participant.
Isolation of peripheral blood mononuclear cells and gut mononuclear cells
Peripheral blood by venipuncture was collected into Acid Citrate Dextran solution A (BD Biosciences) and processed by standard Ficoll–Hypaque density gradient centrifugation to obtain peripheral blood mononuclear cells (PBMCs). Gut biopsies were sampled in the sigmoid colon, at 25 to 30 cm from the anal verge, and collected in RPMI 1640 with 100 U/mL penicillin, 100 μg/mL streptomycin, and 1× GlutaMAX-1 (Invitrogen). The sample was then digested in 0.5 mg/mL clostridiopeptidase A (Sigma) for 30 min at 37°C with shaking, washed, and redigested in 1.0 mg/mL clostridiopeptidase A for an additional 30 min. The resulting cell suspensions were passed through a 70-μm cell strainer (Corning) and washed twice. For in vitro stimulation of T-bet expression in B cells and T-cell–B-cell coculture experiments, frozen PBMCs (in 10% DMSO at −150°C for <2 months) were thawed in complete culture medium (RPMI 1640 + 10% fetal bovine serum [FBS], 10 U/mL penicillin, 10 μg/mL streptomycin, and 1× GlutaMAX-1) supplemented with 0.1% DNase (Sigma), washed twice, and rested overnight in complete medium before use. For all other experiments, fresh PBMCs and gut mononuclear cells were used.
Flow cytometry
For retaining intracellular cytokines, the cells were treated with brefeldin A and monensin (5 μg/mL each) for an additional 6 h at 37°C and 5% CO2; otherwise, cells were stained directly ex vivo or immediately after stimulation/coculture. All staining procedures were performed on ice. Surface antibodies (anti-human CD3, CD4, CD19, IgG, IgM, and IgA; BioLegend) and Violet Dead Cell Stain (Invitrogen) were added in phosphate-buffered saline +2% FBS and incubated with cells for 30 min. The cells were then fixed and permeabilized using the True-Nuclear Transcription Factor Buffer Set (BioLegend) following manufacturer's instructions. Intracellular/nuclear antibodies (anti-human T-bet, IFN-γ, IL-6, IL-10, IL-12, and TNF-α; BioLegend and eBioscience) were added in Perm Buffer (BioLegend) for 30 min before acquisition in FACSCanto cytometer (BD Biosciences).
Quantitative RT-PCR
The cDNA was obtained from 1 g total RNA using Transcriptor First Strand cDNA Synthesis Kit (Roche) following instructions and then analyzed in the ABI PRISM 7700 System (Applied Biosystems). Primers for T-bet, IFN-γ, IL-6, IL-10, IL-12A, IL-17A, and TNF-α were designed using the GenScript Real-time PCR (TaqMan) Primer Design tool. TaqMan GAPDH Control Reagents (Applied Biosystems) was used for normalization.
Cell isolation and coculture
CD4+ T cells and total B cells were isolated using the Human CD4+ T Cell Isolation Kit and Human B Cell Enrichment Kit (Stemcell). The IgG+ and IgG− B cells were sorted by first surface staining purified B cells with fluorescein isothiocyanate (FITC)-conjugated anti-human IgG Fc (clone HP6017) and then using FITC Positive Selection Kit to separate FITC+ and FITC− cells.
Before coculture, CD4+ T cells were first pulsed with anti-CD3 (clone OKT3) antibody for 6 h. The CD4+ T cells were then washed and cocultured with autologous B cells for 48 h. To stimulate B cells, 5 μg/mL anti-B-cell receptor (BCR) (anti-human IgM, clone DA4.4; eBioscience), 1 μg/mL human IFN-γ (Sigma), and/or 1 μg/mL Toll-like receptor 7 (TLR7) agonist (imiquimod; InvivoGen) was added into the cell culture for 48 h. All cells were cultured in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1 × GlutaMAX-1 under 37°C and 5% CO2.
Statistical analyses
All statistical analyses were performed in GraphPad Prism 6 software. Data normality was examined by D'Agostino & Pearson omnibus normality test. Differences between two groups were computed using parametric Student's t-test or nonparametric Mann–Whitney U test accordingly. Differences between three groups were computed using one-way analysis of variance followed by Tukey's (if there was no reference group) or Dunnett's (if there was a reference group) multiple comparison test or Kruskal–Wallis test followed by Dunn's multiple comparison test (if data were not normally distributed). Correlations were examined by Pearson correlation. p < 0.05 is considered significant.
Results
Study participants
Peripheral blood and sigmoid colon samples were obtained from 12 healthy individuals (control), 20 CD patients with active disease (active CD), and 13 CD patients under clinical remission (quiescent CD, CDAI score <150). No treatment was given to active or quiescent CD patients at the time of sample collection. Demographic and disease parameters of all study subjects are summarized in Table 1.
Values are represented as median (range) when applicable.
CD, Crohn's disease; CDAI, Crohn's Disease Activity Index; IBDQ, Inflammatory Bowel Disease Questionnaire; N/A, not applicable.
T-bet-expressing B cells were associated with CD activity
We first examined all study participants for the frequencies of T-bet-expressing B cells (Fig. 1A). In peripheral blood, the frequencies of T-bet+ CD19+ B cells were elevated in active but not quiescent CD patients compared to controls (Fig. 1B). In the gut, the frequencies of T-bet+ CD19+ B cells were elevated in active but not quiescent CD patients. The disease activity, represented by the CDAI, was positively correlated with the frequencies of gut T-bet+ CD19+ B cells in both active CD and quiescent CD patients (Fig. 1C). Together, these results suggested an association between T-bet-expressing B cells and CD development.
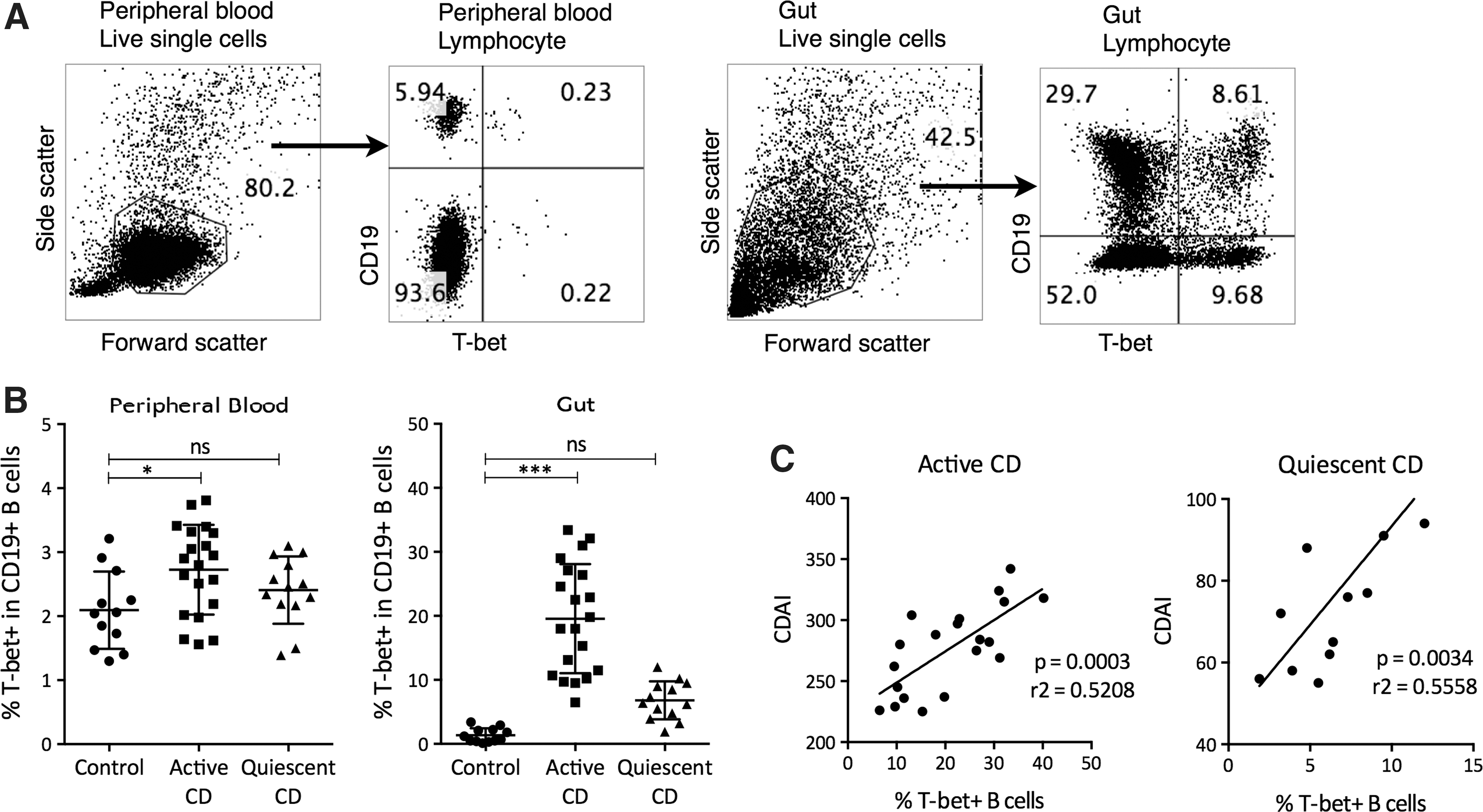
Expression of T-bet in the peripheral blood and gut of CD patients. Peripheral blood mononuclear cells and gut mononuclear cells were stained with anti-human CD19 and Violet Dead Cell Stain for 30 min, fixed and permeabilized for 15 min, and then stained with anti-T-bet for 30 min. The frequencies of T-bet-expressing B cells were examined by flow cytometry.
T-bet-expressing B cells were associated with IgG production and proinflammatory cytokine expression
To examine the functions of T-bet-expressing B cells, we performed flow cytometry staining of antibody secretion and intracellular cytokine production. We found that gut T-bet+ B cells were almost exclusively IgG+, with very few IgM+ and IgA+ cells, while the gut T-bet− B cells were mostly IgM+ or IgA+, with very few IgG+ cells (Fig. 2A, B). B cells also possess inflammatory/regulatory functions independent of their ability to produce antibodies (Lund, 2008). Here, the intracellular cytokine expression was compared between T-bet+ and T-bet− B cells. We found that the T-bet+ B cells contained significantly higher frequencies of IFN-γ+ and IL-6+ cells than the T-bet− B cells (Fig. 2A, C).

Ig isotype and cytokine expression by gut T-bet+ B cells in active CD patients.
Since IgG expression was nearly exclusive to T-bet+ B cells, we used IgG as a surface marker to isolate T-bet+ and T-bet− B cells. Messenger RNA expression of T-bet was exclusively found in IgG+ gut B cells (Fig. 2D). The cytokine production was then quantified by measuring mRNA levels. We found that compared to IgG− gut B cells, IgG+ gut B cells expressed significantly higher levels of IFN-γ, IL-6, and IL-12 (Fig. 2E).
T-bet-expressing B cells contribute to Th1 inflammation
Studies have established that IFN-γ and IL-12 are essential for the initiation and maintenance of Th1 responses, while IL-6 inhibits TGF-β-induced Treg differentiation in favor of Th17 responses. Both Th1 and Th17 contribute to CD pathogenesis (Brand, 2009; Strober and Fuss, 2011). To examine whether T-bet-expressing B cells could mediate CD pathogenesis through promoting Th1 and/or Th17 responses, peripheral blood CD3+CD4+ T cells from active CD patients were cocultured alone or with autologous gut IgG+ or IgG− B cells for 48 h. The CD3+CD4+ T cells were then purified by negative selection, and the cytokine expression was represented by mRNA levels. We found that CD3+CD4+ T cells cocultured with gut IgG+ B cells maintained a higher level of IFN-γ expression than CD3+CD4+ T cells cultured alone (Fig. 3). The CD3+CD4+ T cells cocultured with gut IgG− B cells had reduced level of IFN-γ expression compared to pure CD3+CD4+ T cells. No significant differences were observed in IL-12 and IL-17 expression by CD3+CD4+ T cells.

IgG+ B cells supported Th1 inflammation. Gut B cells (n = 6) from active CD patients were separated into IgG+ and IgG− fractions by FACS sorting. Autologous peripheral blood CD4+ T cells were pulsed with anti-CD3 (0.5 μg/mL) for 6 h and cocultured with IgG+ or IgG− B cells at a 1-to-1 ratio. After 48 h in 37°C and 5% CO2 incubation, the relative IFN-γ, IL-12A, and IL-17A transcript levels were measured by RT-PCR using GAPDH as normalization control. One-way ANOVA followed by Tukey's test. *p < 0.05, ***p < 0.001. Th1, T helper 1.
Previously, T-bet upregulation could be induced in B cells through BCR, TLR7, TLR9, IFN-γ, and/or IL-12 signal transduction pathways. Here, purified peripheral blood B cells from active CD patients were stimulated with IFN-γ, anti-BCR, and TLR7 agonists for 48 h, which significantly elevated the T-bet expression and IFN-γ production by B cells. These B cells (stimulated B) were then washed and cocultured with autologous CD3+CD4+ T cells. Alternatively, CD3+CD4+ T cells were cocultured with unstimulated peripheral blood B cells (unstimulated B). After 48 h, CD3+CD4+ T cells cocultured with stimulated B had significantly higher IFN-γ and IL-12 production compared to those cocultured with unstimulated B (Fig. 4C). Together, these studies suggested that T-bet-expressing B cells supported Th1-type inflammation.
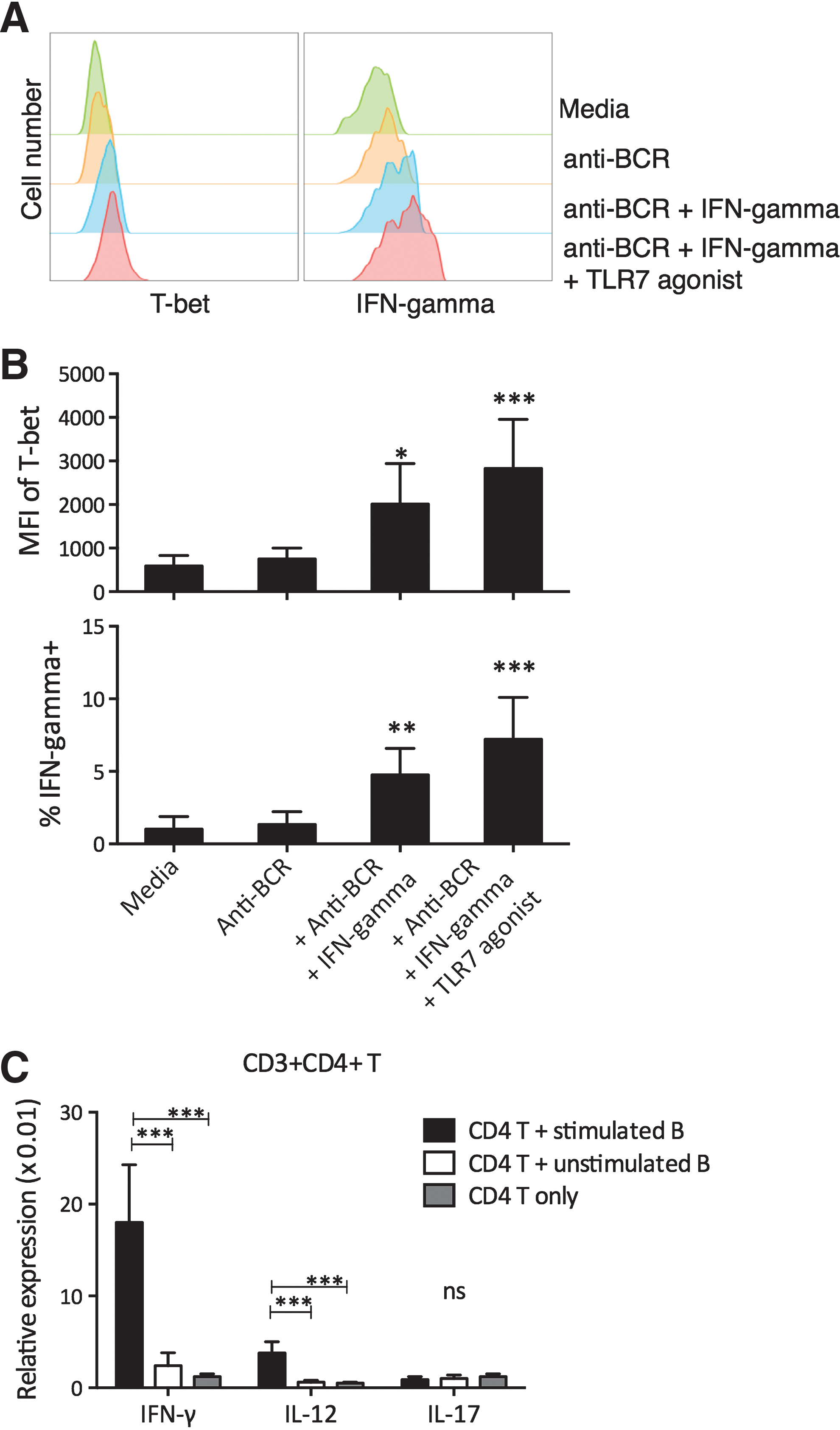
In vitro stimulation of T-bet expression in B cells and support of Th1 inflammation. Purified peripheral blood B cells from active CD patients were stimulated with combinations of anti-human BCR (anti-IgM, DA4.4), IFN-γ, and TLR7 agonist (imiquimod) for 48 h.
T-bet-expressing B cells are associated with Th1 infiltration in the gut epithelium
Based on these results, we suspected that gut T-bet-expressing B cells could exacerbate Th1 inflammation in the gut, thus directly contributing to CD development. To examine this, the active CD patients were grouped into T-bet+ B-high and T-bet− B-low groups using the median gut T-bet+ B-cell percentage (18.9%) as a cutoff. We found that T-bet+ B-high patients had significantly higher levels of gut-infiltrating Th1 cells, identified by IFN-γ and IL-12 production, than T-bet− B-low patients (Fig. 5).
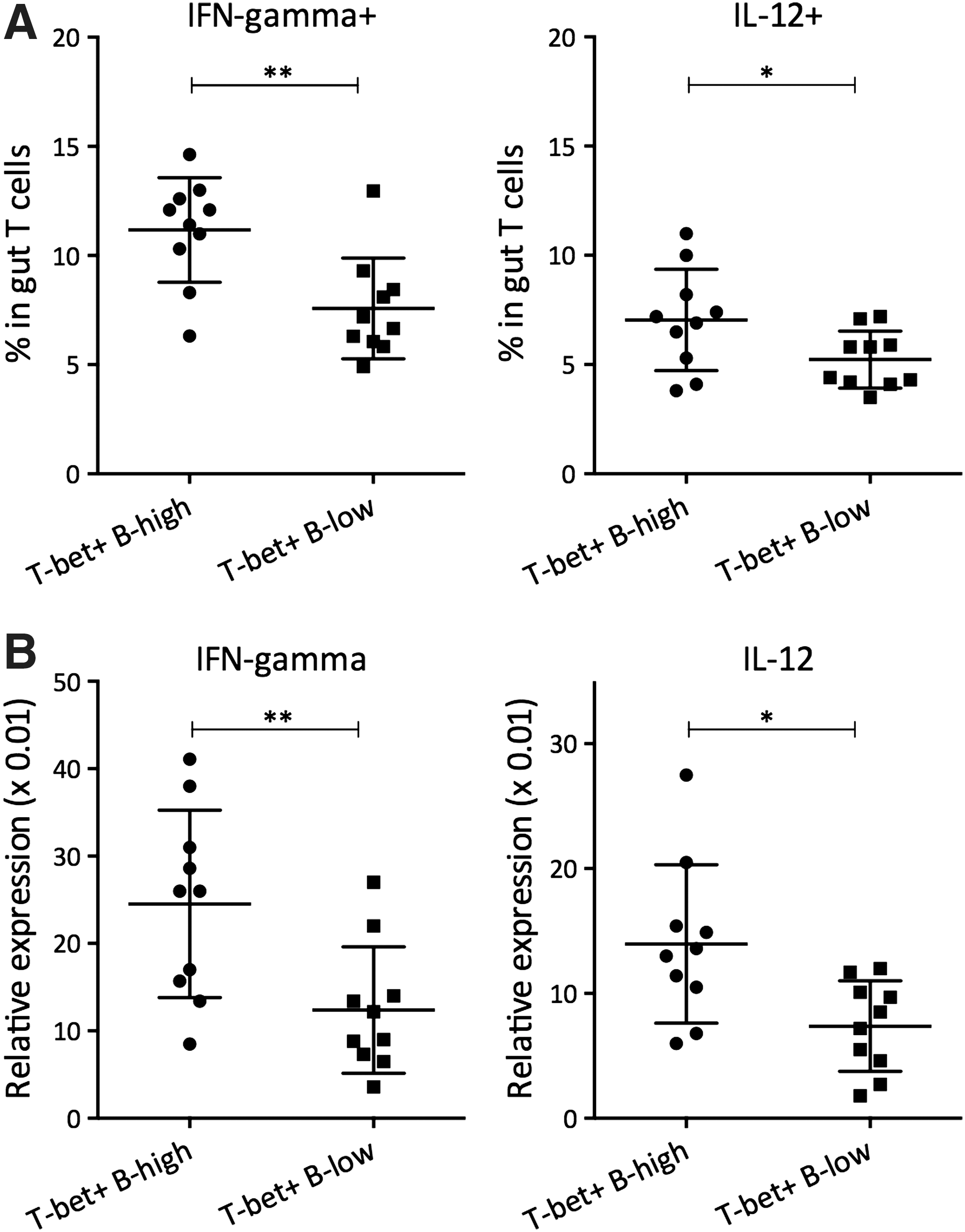
Gut Th1 inflammation in T-bet+ B-high versus T-bet+ B-low patients.
Discussion
In this report, we characterized the role of T-bet-expressing B cells in human CD patients. We first discovered an association between T-bet-expressing B-cell enrichment and CD disease status. Active, but not quiescent, CD patients presented an elevated frequency of T-bet-expressing B cells in the peripheral blood and strongly in the gut. Moreover, the frequency of T-bet-expressing B cells in the gut was directly correlated with the CDAI score, suggesting an association between gut T-bet-expressing B cells and disease activity. Whether T-bet-expressing B cells can act as a drug target requires further investigations. In addition, to the best of our knowledge, the upregulation of T-bet in CD patients is not entirely clear. Current studies have largely attributed T-bet induction to IFN-γ-mediated signal transduction, concomitant with BCR and TLR activations (Harris et al., 2005; Lazarevic et al., 2013). It is unclear whether T-bet can be upregulated in B cells directly by intestinal injuries and insults, by particular types of microorganisms, or requiring Th1 inflammation in advance.
Surface and intracellular staining showed that T-bet+ B cells were predominantly IgG+ and expressed higher levels of IFN-γ, IL-6, and IL-12 than T-bet− B cells. To examine whether these T-bet-expressing B cells supported or inhibited Th1 and Th17 inflammation, we used IgG+ expression to isolate T-bet-expressing B cells, which were cocultured with autologous blood CD4+ T cells. We found that gut IgG+, but not IgG−, B cells supported IFN-γ production from autologous blood CD4+ T cells. Due to the scarcity of gut B cells, we induced T-bet expression in peripheral blood B cells through stimulation of BCR, TLR7, and IFN-γ, which resembled gut T-bet+ B cells in terms of elevated IFN-γ. We found that these stimulated B cells, but not unstimulated B cells, supported the IFN-γ and IL-12 production from autologous CD4+ T cells. Together, these results suggest that T-bet-expressing B cells in CD may exacerbate CD disease activity through supporting Th1 inflammation. Moreover, previous studies indicated that IFN-γ was indispensable in orchestrating intestinal inflammation with Th17 cells, but T-bet- and IFN-γ-expressing Th1 cells were not required (Simpson et al., 1998; Gökmen et al., 2013; Zimmermann et al., 2016). These results showed that abundant amount of T-bet-expressing B cells were present in the intestinal tract and capable of supplying IFN-γ, thus suggesting a contributing role of T-bet-expressing B cells in CD inflammation. In addition, we identified that T-bet-expressing B cells were major IgG producers in the gut (Fig. 2B). Binding of IgG on cell surface and recognition of IgG Fc by the NK cell CD16 Fc receptor can lead to abundant production of IFN-γ in antibody-dependent cellular cytotoxicity (Seidel et al., 2013). Whether this mechanism contributes to CD inflammation requires further investigations.
We also demonstrated a positive association between the frequencies of T-bet-expressing B cells and the percentage of gut-infiltrating IFN-γ+ and IL-12+ CD4+ T cells in active CD patients. Further studies using well-controlled animal models and adoptive transfers are required to examine whether T-bet-expressing B cells assist the differentiation, maintenance, and infiltrating of Th1 cells in the gut. In addition, whether T-bet-expressing B cells could affect CD patients’ responses to treatment, risk of relapse, and disease prognosis requires further examinations.
The B-cell-mediated inflammation is being increasingly recognized and has been shown to play critical roles in the induction and resolution of autoimmunity and other inflammatory disorders (Serreze et al., 1996; Matsushita et al., 2008; Liu et al., 2014). In the intestinal tract, the B cells/plasmablasts were previously ascribed with regulatory activity due to their expression of IL-10 and were thought to suppress CD4+ and CD8+ T-cell function, including IFN-γ expression (Mauri and Bosma, 2012; Liu et al., 2014; Matsumoto et al., 2014). Here, we observed that intestinal T-bet+ and T-bet− B cells secreted comparable levels of IL-10 (Fig. 2C, D), but T-bet+ B cells had promoted, rather than suppressed, IFN-γ expression by T cells (Fig. 4). This result is consistent with other studies that showed IL-10 in collaboration with IFN-γ might promote T-cell inflammation by increasing cell survival and proliferation (Emmerich et al., 2012; Oft, 2014). It also highlighted the complexity of B-cell-mediated regulations. In type 1 diabetes, IL-10-expressing regulatory B cells contributed to the maintenance of tolerance to islet autoantigens, but depletion of total B cells enabled the reduction of immune response against islet peptides in vitro and increased the presence of regulatory T cells in vivo (Fiorina et al., 2008; Kleffel et al., 2015). B cell targeting via TIM4 favored Th2 instead of Th1 inflammation (Vergani et al., 2015). This study, together with previous ones, demonstrated the existence of multiple B-cell subsets with distinctive roles in immune regulation.
Footnotes
Acknowledgment
We thank Dr. Song at the DICAT Biomedical Computational Centre for his technical support.
Disclosure Statement
No competing financial interests exist.