
Abstract
Lysyl oxidase (LOX) is a copper-dependent enzyme that catalyzes covalent cross-linking of collagen. In response to hypoxia, phosphatidylinositol 3-kinase (PI3K) pathway is activated and contributes to pulmonary arterial hypertension (PAH). However, potential role of LOX in hypoxia-induced PAH is poorly understood. In this study, we explored the mechanism responsible for the development of hypoxia-induced PAH. Potent inhibitors of PI3K/Akt and LOX, wortmannin and β-aminopropionitrile (β-APN), were administrated in rat model of hypoxia-induced PAH. The cross-linking of collagen was assessed by the determination of hydroxyproline. LOX, LOXL-1, LOXL-2, LOXL-3, LOXL-4, Akt, and phospho-Akt expression was detected by real-time polymerase chain reaction and western blot analysis. We observed that collagen cross-linking and LOX activity were elevated in hypoxia-exposed rat lung tissue, but these effects were reversed by β-APN and wortmannin. In addition, exposure to hypoxia enhanced mRNA and protein expression and activity of LOX and LOXL-1 in a PI3K/Akt-dependent manner and induced the development of PAH. After the administration of wortmannin, the upregulation of LOX and cross-linking of collagen were significantly reversed in hypoxia-exposed rat pulmonary artery tissue. Taken together, the present study demonstrated that the upregulation of LOX expression and collagen cross-linking is PI3K/Akt dependent in rat with hypoxia-induced PAH. Suppression of PI3K/Akt pathway may alleviate hypoxia-induced PAH through the downregulation of LOX.
Introduction
H
It was demonstrated that the stiffness of pulmonary artery was involved in the deposition and cross-linking of extracellular matrix (ECM) protein collagen and elastin (Wang and Chesler, 2012). The covalent cross-linking of these ECM proteins catalyzed by lysyl oxidase (LOX) is crucial to the formation of insoluble collagen and elastic fiber. LOX is a copper-dependent amine oxidase, and the family of LOXs includes LOX and four LOX-like proteins (LOXL-1, -2, -3, and -4). LOX is initially synthesized in vascular smooth muscle cells (VSMCs) and fibroblasts as a 46 kDa preproenzyme, which is secreted into ECM and further processed by proteolysis into a 32 kDa active form (Rodríguez et al., 2008).
Recently, several growth factors and cytokines have been shown to regulate LOX expression and activity. While transforming growth factor-β (TGF-β), hypoxia-inducible factor-1α (HIF-1α), and platelet-derived growth factor (PDGF) increase LOX expression and activity, tumor necrosis factor-α reduces LOX expression and activity in endothelial cells and in the vascular wall (Green et al., 1995; Atsawasuwan et al., 2008; Rodríguez et al., 2008; Schietke et al., 2010; Pez et al., 2011). Of note, hypoxia-induced upregulation of TGF-β, HIF-1α, and PDGF plays a pivotal role in PAH (Semenza, 2005; Thébaud et al., 2005; Selimovic et al., 2009; Gong et al., 2011; Chen et al., 2014). Growth factors that are elevated in vessels such as TGF-β, HIF-1α, and PDGF activate phosphatidylinositol 3-kinase (PI3K)/Akt signaling in smooth muscular cells (SMCs) (Heldin et al., 1998; Goncharova et al., 2002; Stewart et al., 2008). Furthermore, activation of PI3K/Akt promoted SMC proliferation, while Akt inhibitor LY294002 attenuated hypoxia-induced PAH (Garat et al., 2013). Interestingly, the induction of LOX protein in response to TGF-β could be prevented by the inhibitor of PI3K wortmannin (Voloshenyuk et al., 2011). In contrast, under hypoxic condition LOX may activate PI3K/Akt pathway, thereby promoting tumor cell proliferation (Schietke et al., 2010).
Therefore, in this study we investigated the interaction between PI3K/Akt and LOX and explored potential mechanism responsible for the development of hypoxia-induced PAH. Using LOX inhibitor β-aminopropionitrile (β-APN) and PI3K inhibitor wortmannin, we provided the evidence that the upregulation of LOX expression induces collagen cross-linking in hypoxia-induced rat pulmonary artery partly by involving PI3K/Akt signaling.
Materials and Methods
Chronic hypoxia-induced rat model of PAH
Pathogen-free male Sprague-Dawley rats (weight 200–250 g) were obtained from Animal Laboratory Center of Wenzhou Medical University and maintained in a capsule for 8 h per day and 6 days per week for up to 3 weeks. All animal protocols were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University. Rats were maintained in a capsule with hypoxic air (10% O2 and 90% N2) for 8 h per day and 6 days per week for up to 3 weeks. Rats were then administrated with intraperitoneal injection of wortmannin (15 μg/kg/day in dimethyl sulfoxide [DMSO]; Biolmol), β-APN (100 mg/kg/day in normal saline; Sigma-Aldrich), or vehicle (DMSO or normal saline) as previously described (Singh et al., 2001; Wang et al., 2012) and then exposed to hypoxia. As per the control, rats in the normoxic condition were injected with normal saline and exposed to room air. At the end of the 21-day exposure period, all rats were weighted and intraperitoneally anesthetized with pentobarbital injection (30 mg/kg). Then the right neck external carotid artery and jugular vein were dissected.
To monitor mean pulmonary artery pressure (mPAP), a catheter pressure transducer (Shanghai Medical Electronic Instrument) was inserted into the main pulmonary artery through right jugular vein and right ventricle (RV). After hemodynamic measurement, rat was killed by exsanguination. Then the chest cavity was opened, and the lung and heart were quickly separated. For the assessment of right ventricular hypertrophy, the RV and left ventricle (LV) plus septum were dissected and weighted. The ratio of RV to LV plus septum weight (RV/LV + S) was calculated and used as an indirect indicator of PAH and RV hypertrophy.
Histopathological analysis
Right lung tissue was cut as cross sections ∼5 mm × 5 mm × 5 mm in size, then fixed in 4% paraformaldehyde for 24 h at 4°C, embedded in paraffin, and sectioned into 5 μm thickness. For histological analysis, sections were stained with hematoxylin-eosin (HE) and examined under light microscopy. To evaluate pulmonary arterial remodeling, the percentage of medial thickness of pulmonary artery was analyzed by the following formula: the percentage of medial thickness = wall areas/total areas.
Assay of collagen cross-linking
To distinguish between cross-linking (insoluble) and noncross-linking (soluble) collagen, a colorimetric procedure was used. The pepsin-soluble collagen in lung tissue was extracted with 5 mg/mL pepsin in 0.5 M acetic acid overnight. The soluble and insoluble collagens were separated by centrifugation at 2000 g for 6 min at 4°C. The total soluble and insoluble collagens were assessed based on the determination of hydroxyproline according to the manufacturer's instructions (Nanjing Jiancheng). The degree of cross-linking was calculated as the ratio between the insoluble and soluble forms of collagen.
LOX activity assay
LOX enzyme activity was measured using the Fluorimetric Lysyl Oxidase Assay Kit (AAT Bioquest) according to the manufacturer's instructions. Briefly, lung tissue was homogenized in extraction buffer containing 1.2 mM urea and 50 mM sodium borate (pH 8.2) at 4°C overnight. After centrifugation, supernatant was collected and incubated with a reaction mixture containing 50 mM sodium borate, 1.2 mM urea, pH 8.2, 1 U/mL horseradish peroxidase, 50 μM Amplex Red, and 10 mM 1.5-diaminopentane with or without 500 μM β-APN (Sigma) at 37°C for 30 min. LOX activity was assayed by monitoring LOX-catalyzed H2O2 release from the fluorescent substrate. The fluorescent product was excited at 560 nm, and the emission was read at 590 nm using a fluorescence spectrophotometer (Hitachi Instruments). All enzyme activities were expressed as fluorescence at 590 nm. LOX activity was calculated as the increase in fluorescent units.
Quantitative real-time polymerase chain reaction analysis
Total RNA was extracted from pulmonary artery tissue with TRIzol (Invitrogen Life Technologies) and purified with RNeasy Mini Kit columns (QIAGEN). cDNA was synthesized from 2 μg of purified RNA using reverse transcriptase (RevertAid First Strand cDNA Synthesis Kit; Fermentas), according to the manufacturer's instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was carried out on an ABI PRISM 7300 PCR Detection System.
The sequences of the primers were as follows: LOX forward 5′-GCCTGGACCGTGCTCTTTCT-3′, reverse 5′-CGTTGTTCTCCCATTGGATTGT-3′; LOXL-1 forward 5′-ACTTGCCTGTGCGAAACTCT-3′, reverse 5′-CCTGCACGTAGTTGGGATCT-3′; LOXL-2 forward 5′-ATAATGAAGGCCGTGTGGAG-3′, reverse 5′-AGTGACACCCCAGCCATTAG-3′; LOXL-3 forward 5′-CAGGACCAGGACTCTGCTTC-3′, reverse 5′-CCATACTGGAGCTGCACAGA-3′; LOXL-4 forward 5′-GACAAGGAGCACCAAGAAGC-3′, reverse 5′-TTCATCCCTGAGGAACTTGG-3′; and β-actin forward 5′-GACATAAAGGAGAAGCTGTGC-3′, reverse 5′-CATGATGGAGTTGAAGGTGGT-3′. The expected size of PCR product was 127 bp for LOX, 150 bp for LOXL-1, 239 bp for LOXL-2, 152 bp for LOXL-3, 229bp for LOXL-4, and 219 bp for β-actin. Amplification was performed at 50°C for 3 min, 95°C for 15 min, followed by 40 cycles at 95°C for 10 s, 61.5°C for 20 s, and extension at 72°C for 20 s. cDNA products were used as templates for qRT-PCR to measure gene expression levels using a SYBR Green System (SuperReal PreMix Plus; TIANGEN). Standard curves were plotted for each target gene and internal control (β-actin). Data were calculated using the 2−ΔΔCt method, and relative mRNA expression was normalized to β-actin mRNA.
Western blot analysis
Pulmonary artery tissue was homogenized for 1 h at 4°C in a solution containing 50 mM Tris HCl, 1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM NaF, 1 mM phenylmethane sulfonyl fluoride (PMSF), 1 mM Na3VO4, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate (pH 7.4), and protease inhibitor cocktail (Pierce Biotechnology). Samples were centrifuged at 12,000 g for 15 min, and supernatants were collected. Protein concentration was assessed using the Pierce BCA Protein Assay Kit (Pierce Biotechnology). Proteins were loaded on SDS-polyacrylamide gel electrophoresis gels (Invitrogen) and blotted electrophoretically onto nitrocellulose membranes (Invitrogen). The membranes were blotted with a 4% dry milk powder in 0.2% Tween 20-phosphate-buffered saline buffer at room temperature for 1 h and then incubated with rabbit polyclonal primary antibodies for LOX, LOXL-1, LOXL-2, LOXL-3, LOXL-4 (1:400; Abcam), Akt (1:500; Abcam), Phospho-Akt (p-Akt, 1:200 Ser473; Abcam), and β-actin (1:5000; Abcam) at 4°C overnight. After washing thrice, membranes were incubated with secondary antibody conjugated with horseradish peroxidase (Abcam) at room temperature for 2 h. Bands were digitized by ImageQuant software (Bio-Rad) and quantified by densitometry with normalization to β-actin.
Statistical analysis
Statistical differences among many groups were performed by One Way analysis of variance followed by post hoc comparison using Sigma Plot software. All data were expressed as mean ± standard error of the mean (SEM) and p < 0.05 indicated significant difference.
Results
LOX or PI3K inhibitor blocks hypoxia-induced hypertension, RV hypertrophy, and pulmonary arterial remodeling
Compared to normoxia-exposed rats, 3-week exposure to hypoxia led to significant increases in mPAP and the ratio of RV to LV plus septum weight (RV/LV + S) (Fig. 1). However, after rats exposed to hypoxia were concurrently administrated with LOX inhibitor β-APN or PI3K inhibitor wortmannin, hypoxia-induced increases in mPAP and RV/LV + S were significantly reduced, but did not reach the normoxic levels (Fig. 1).

The mPAP and the ratio of RV to LV plus septum (RV/LV + S) in different groups of rats.
Furthermore, HE staining revealed that the percentage of wall thickness of pulmonary artery in hypoxia-exposed rats was significantly elevated compared with normoxia-exposed rats (Fig. 2A, B). However, upon administration with either β-APN or wortmannin, the elevated wall thickness of pulmonary artery in hypoxia-exposed rats was inhibited (Fig. 2C, D).
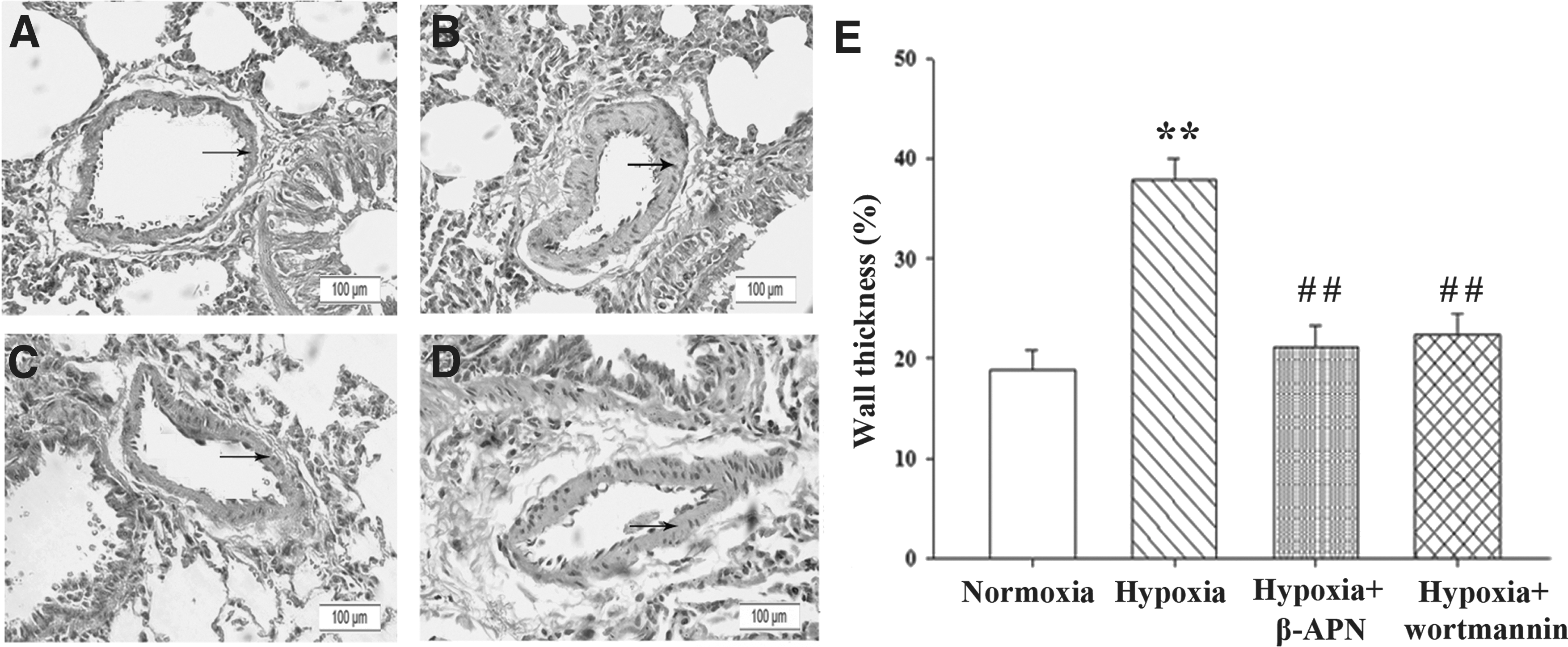
Wortmannin inhibits hypoxia-induced pulmonary vascular remodeling. Hematoxylin-eosin staining showed wall thickness of pulmonary artery in the lung from normoxic
LOX or PI3K inhibitor attenuates collagen cross-linking in lung tissue of hypoxia-exposed rats
The contents of both insoluble and soluble collagen in lung tissue of hypoxia-exposed rats were significantly elevated compared with normoxia-exposed rats. In addition, the ratio of insoluble collagen to total collagen was lower in lung tissue of normoxic rat, but was obviously increased after exposure to hypoxia. However, the higher contents of collagen and collagen cross-linking in lung tissue of hypoxia-exposed rats were significantly decreased upon administration with either β-APN or wortmannin (Table 1).
Values shown are mean ± standard error of the mean from eight independent experiments. ** p < 0.01 vs. normoxia; ## p < 0.01 vs. hypoxia.
β-APN, β-aminopropionitrile.
PI3K inhibitor decreases LOX activity in lung tissue of hypoxia-exposed rats
As shown in Figure 3A, there was 3.68-fold increase in LOX activity of hypoxic rat lung tissue compared to that of normoxic rat. Interestingly, upon administration with wortmannin, there was 2.53-fold decrease in LOX activity of hypoxic rat lung tissue compared to control (Fig. 3A).

Wortmannin inhibits hypoxia-induced upregulation of LOX activity and mRNA expression in pulmonary artery tissue.
LOX expression is upregulated in hypoxia-exposed rat pulmonary artery tissue
Next we wondered whether hypoxic exposure is able to affect LOX expression at mRNA and protein levels. Real-time PCR showed that there were significant elevations of LOX and LOXL-1 mRNA levels, but not LOXL-2, LOXL-3, or LOX-4 in hypoxia-exposed rat pulmonary artery tissue compared with normoxia-exposed tissue (Fig. 3B). Moreover, western blot analysis revealed that exposure to hypoxia led to the upregulation of LOX and LOXL-1 protein levels compared with normoxia-exposed tissue (Fig. 4A, B). Taken together, these data indicate that LOX and LOXL-1 are induced in rat pulmonary artery tissue after the exposure to hypoxia.
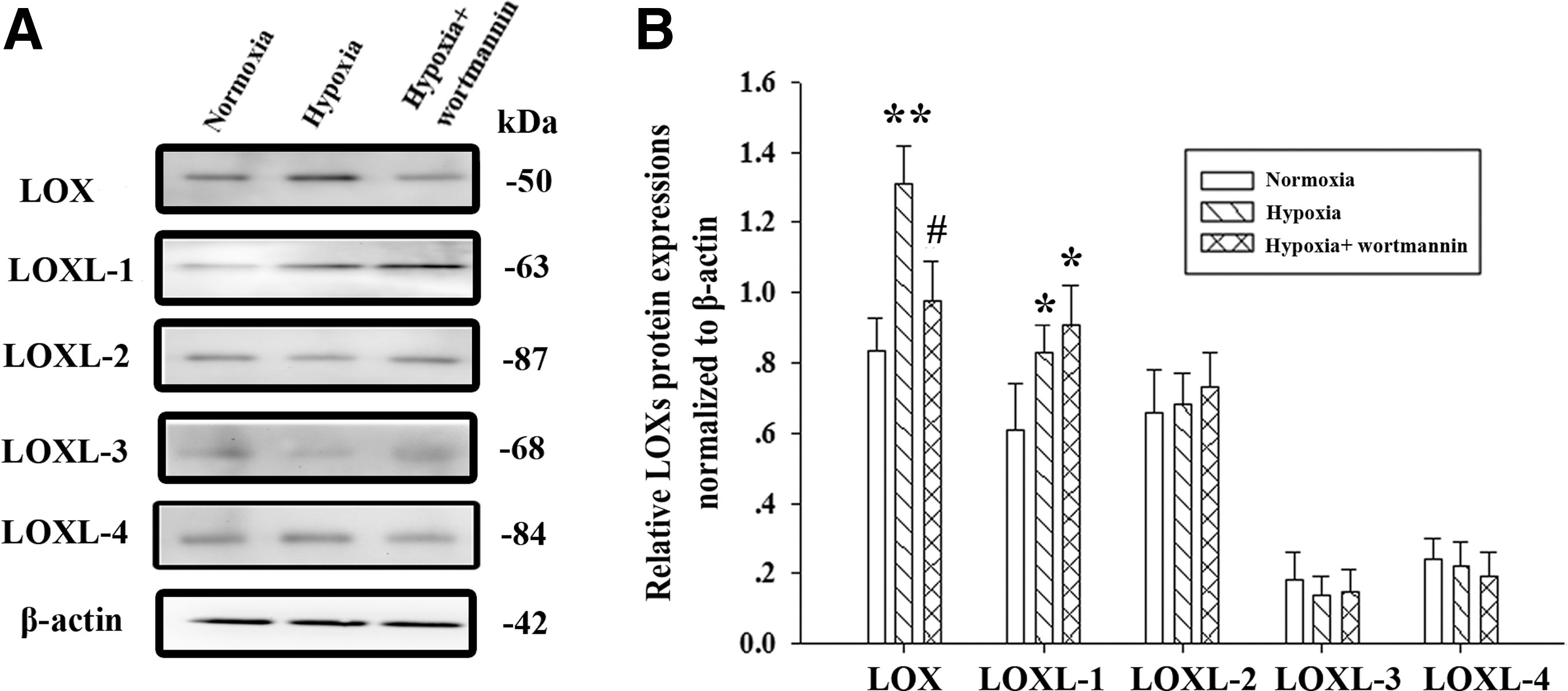
Wortmannin inhibits hypoxia-induced upregulation of LOX protein in pulmonary artery tissue.
PI3K/Akt signaling regulates LOX expression in hypoxia-exposed rat pulmonary artery tissue
To investigate the effect of hypoxia on PI3K/Akt signaling, we performed western blot analysis of rat pulmonary artery tissue and found that hypoxia led to the upregulation of Akt (Fig. 5A, C) and p-Akt (Fig. 5B, D) in rat pulmonary artery tissue. As expected, administration with wortmannin reduced the ratio of p-Akt/Akt in hypoxia-induced rat pulmonary artery tissue (Fig. 5E).
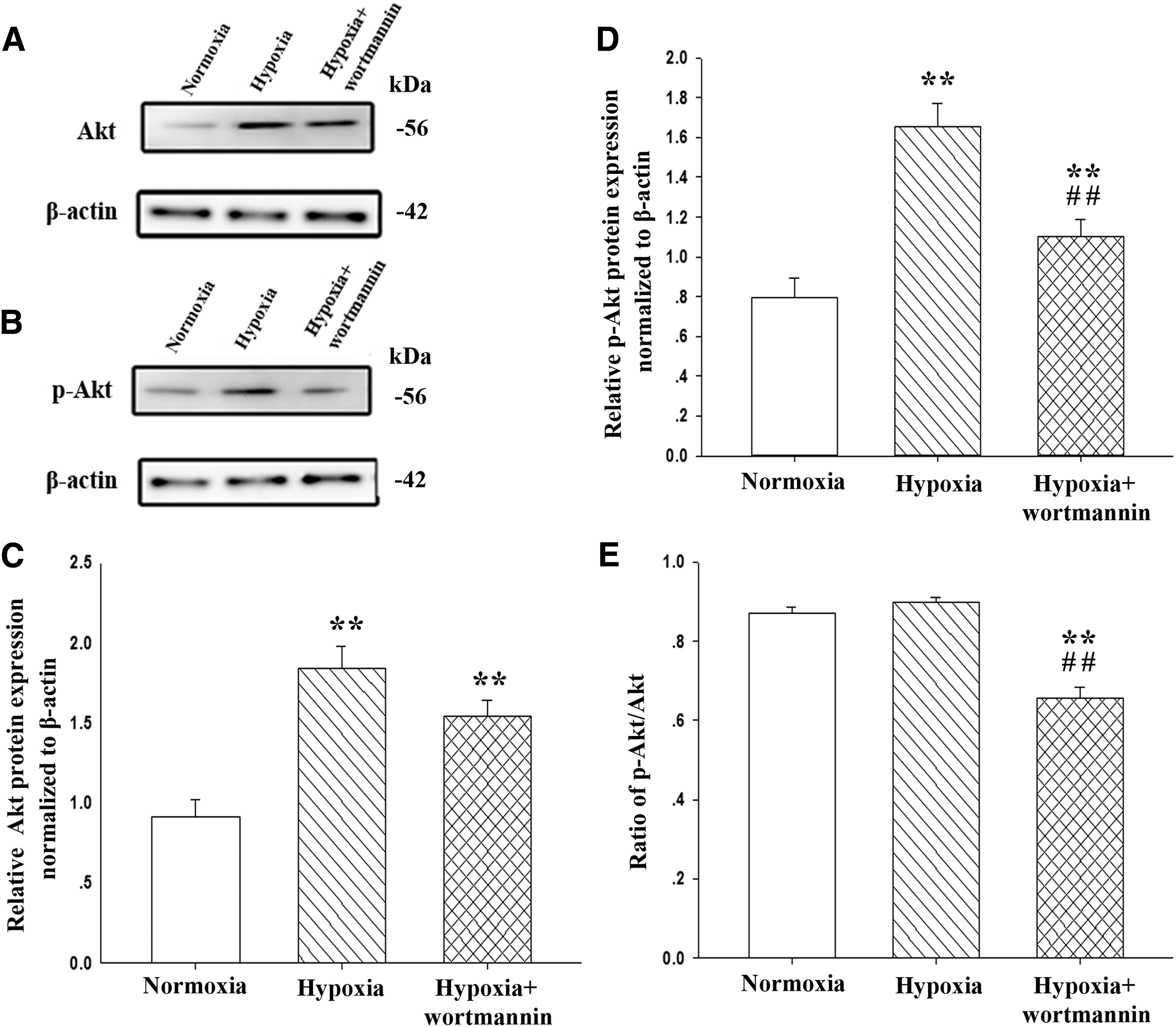
Wortmannin inhibits hypoxia-induced activation of Akt in pulmonary artery tissue. Western blot analysis of Akt
To understand whether PI3K/Akt is involved in the regulation of LOX, we applied wortmannin to inhibit PI3K/Akt signaling. Consistent with a decrease in Akt phosphorylation upon administration with wortmannin (Fig. 5B, D, and E), mRNA and protein levels of LOX, but not LOXL-1, LOXL-2, LOXL-3, or LOX-4 were significantly reduced in hypoxia-exposed rat pulmonary artery tissue (Figs. 3B and 4A, B).
Discussion
Hypoxia is the primary trigger that contributes to the development of PAH and pulmonary arterial remodeling. In this study, our data showed that the upregulation of LOX and LOXL-1 was involved in increased cross-linking of collagen in hypoxia-exposed rat lung tissue of PAH, but this could be partly attenuated by the administration of LOX inhibitor β-ANP. Furthermore, our results indicated that hypoxia-induced upregulation of LOX, but not LOXL-1, LOXL-2, LOXL-3, or LOX-4 at mRNA and protein levels may be mediated by PI3K/Akt signaling because PI3K inhibitor wortmannin significantly reversed the upregulation of LOX and decreased the cross-linking of collagen and the development of PAH.
ECM in the lung is composed of two major structural proteins, collagen and elastin, which are synthesized and secreted by fibroblasts and myofibroblasts. The balance of ECM cross-linking is important for sustaining vascular tension and stiffness. Excessive cross-linking of collagen was confirmed to play an important role in arterial remodeling after balloon injury (Brasselet et al., 2005). In addition, a transgenic mouse model study suggested that abnormal accumulation of collagen was involved in hypoxia-induced PAH (Wang et al., 2012). Furthermore, over cross-linking of collagen was shown in hypoxia-induced pulmonary arterial hypertensive mice (Nave et al., 2014). Consistent with these previous studies, our present study revealed that there were significant increases in both content and cross-linking of collagen in hypoxia-induced rat lung tissue. Thus, we speculate that increased content and cross-linking of collagen contribute to the development of hypoxia-induced PAH (Wang et al., 2012).
Acting as the key copper-dependent amine oxidase, LOX catalyzes the covalent cross-linking in elastin and collagen. LOX, LOXL-1, 2, 3, and 4 are ubiquitously distributed in the lung, heart, skeletal muscle, kidney, and uterus. However, under normal condition, the expression of LOX, LOXL-1, LOXL-2, LOXL-3, and LOXL-4 appears to be low in most tissues. In the vascular wall, LOX is expressed in fibroblasts, endothelial cells, and VSMCs. LOX has been associated with VSMC migration and proliferation, mainly because LOX is the isoform responsible for 80% of LOXs activity in VSMC (Rodríguez et al., 2008). Notably, hypoxia has been shown to upregulate LOX expression in many cancer cell lines (Payne et al., 2005; Erler et al., 2006; Ji et al., 2013). In the present study, we further observed that LOX and LOXL-1 expression at either mRNA or protein level was upregulated, and their activity was elevated in hypoxia-exposed rat pulmonary artery tissue. However, no effects on the mRNA and protein levels of LOXL-2, LOXL-3, or LOXL-4 were observed in rat pulmonary artery tissue after exposure to hypoxia. These data suggest that LOX and LOXL-1, but not LOXL-2, LOXL-3, or LOXL-4 could be induced by hypoxia. After the administration of β-ANP to suppress LOX, elevated collagen cross-linking was reversed and pulmonary arterial pressure was decreased. Our findings are in agreement with those previously described by Nave et al. (2014) that overexpression of LOX could be crucial for hypoxia-induced ECM cross-linking and the development of PAH.
Hypoxia is an initial culprit for the development of PAH. Multiple mechanisms may be involved in hypoxia-mediated LOX production. Under hypoxic condition, HIF-1α is induced, which may upregulate LOX expression (Ji et al., 2013). Furthermore, growth factors such as TGF-β and PDGF are critical to the induction of LOX expression in response to hypoxia and contribute to VSMC growth (Rodríguez et al., 2008). Conversely, basic fibroblast growth factor and Interferon-γ downregulate LOX expression in rat aortic smooth muscle cells (Song et al., 2000). Interestingly, hypoxia-induced HIF-1α expression could lead to the activation of PI3K and Akt (Schietke et al., 2010). In addition, several reports revealed that hypoxia triggers PDGF to activate PI3K/Akt signaling and promote VSMC proliferation and pulmonary artery thickening (Gao et al., 2006; Garat et al., 2006).
Previous studies reported that PI3K/Akt inhibitor decreased the proliferation of pulmonary artery smooth muscle cells and pulmonary vascular endothelial dysfunction under hypoxic condition (Liu and Fanburg, 2006; Li et al., 2011). In present study, we observed that Akt and p-Akt were upregulated in hypoxia-induced pulmonary hypertensive rat pulmonary artery tissue. Although there was no significant change in the ratio of p-Akt/Akt between hypoxic and normoxic pulmonary artery tissue, the administration of wortmannin significantly reduced the ratio of p-Akt/Akt. Furthermore, our results demonstrated that the inhibition of PI3K/Akt by wortmannin effectively downregulated LOX expression at protein and mRNA levels and reversed the elevated cross-linking of collagen and pulmonary arterial pressure in hypoxia-exposed rats, thus partly reversing hypoxia-induced PAH. Taken together, these observations demonstrate that PI3K/Akt signaling plays a pivotal role in the development of PAH and the remodeling of pulmonary artery smooth muscle.
In conclusion, the present study provides evidence that hypoxia leads to the upregulation of LOX expression and the elevation of collagen cross-linking through the activation of PI3K/Akt signaling. Suppression of PI3K-LOX pathway may contribute to the improvement of hypoxia-induced PAH. Therefore, therapies that target PI3K/Akt and LOX are potentially novel approach for the treatment of PAH.
Footnotes
Acknowledgment
This study was supported by a grant from Zhejiang Province Natural Science Foundation of China (No. LY12H01002).
Disclosure Statement
No competing financial interests exist.