
Abstract
Oxidative stress is well known to play a pivotal role in hypoxia/reoxygenation (H/R)-induced neuron injury. On the basis of this fact, antioxidative agents have been demonstrated to be neuroprotective. 17-DMAG (HSP90 inhibitor) is reported to have neuroprotective effects in vitro, which may interfere with oxidative stress through reduction in pro-oxidative factors. However, little is known about its effects on H/R-induced neuron injury and the underlying mechanisms. In this study, the effects of 17-DMAG on H/R-treated HT22 cells were investigated. MTT and lactate dehydrogenase (LDH) assays indicated that 17-DMAG led to a dose-dependent recovery of cell viability in H/R-treated HT22 cells. Flow cytometry demonstrated that 17-DMAG inhibited the cell apoptosis induced by H/R in HT22 cells. In addition, Western blot and real-time reverse transcription–polymerase chain reaction indicated that 17-DMAG inhibited the H/R-induced upregulation of Bax/Bcl-2 ratio and cleaved caspase-3 expression. Moreover, our results demonstrated that 17-DMAG promoted the expression of antioxidant enzymes, including manganese superoxide dismutase, catalase, and glutathione peroxidase. As a result, 17-DMAG might resist to H/R-induced oxidative stress. Furthermore, 17-DMAG increased the expression of phosphorylation of Akt (p-Akt) and the heme oxygenase-1 (HO-1), as well as the translocation of nuclear factor erythroid 2-related factor 2 (Nrf2) in H/R-treated HT22 cells. However, the Akt inhibitor, LY294002, partially hampered the effects of 17-DMAG on the expression of p-Akt, nuclear Nrf2, and HO-1 and cell viability, as well as cell apoptosis induced by H/R in HT22 cells. In conclusion, the findings of our study thus demonstrate that 17-DMAG protects against H/R-induced HT22 cell injury through Akt/Nrf2/HO-1 pathway, which may be associated with its antiapoptotic and antioxidative stress effects.
Introduction
C
Heat shock protein 90 (HSP90) is a ubiquitous molecular chaperone that ensures the proper conformation of proteins, including key mediators of signal transduction and transcriptional regulation (Pratt and Toft, 2003). Selective HSP90 inhibitors 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG) block the ATP-binding site of HSP90 and exert pleiotropic functions, which include induction of the heat-shock response (Zhang and Burrows, 2004). Although the 17-DMAG is of therapeutic interest primarily in cancer (Liu et al., 2016; Zhang et al., 2016), evidence is emerging for the potential beneficial role of HSP90 inhibitors in the treatment of other inflammatory diseases, including ischemic stroke (Lu et al., 2002). Also, 17-DMAG has been reported to protect blood–brain barrier integrity in the MCAO-induced C57/BL6 mouse model and OGD/reoxygenation-induced bEnd.3 cells (Qi et al., 2015). A previous study reported that 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration (Tokui et al., 2009). However, the protective effects of 17-DMAG have little been examined in neurodegenerative diseases in vitro.
Oxidative stress is known to have an important role in the pathogenesis of ischemic CVD (De Silva and Miller, 2016). The neuronal cell death observed in individuals after acute injury or in those with a chronic disease is frequently caused by oxidative stress. Hypoxia/reoxygenation (H/R) can cause oxidative stress in a process that involves the formation of reactive oxygen species (ROS). Overproduction of ROS and a diminished antioxidant defense system have been linked to enhanced oxidative stress state in the CVD (De Silva and Miller, 2016). Endogenous antioxidant systems include a number of proteins or enzymes, for example, manganese superoxide dismutase (MnSOD), catalase (CAT), and glutathione peroxidase (GPx), as well as nonenzymatic, low-molecular-weight antioxidant compounds, which constitute the first line of defense against oxidative stress and maintain the redox environment of the body (Armogida et al., 2012). A previous study has recently reported that 17-DMAG diminishes the inflammatory response in atherosclerosis through inhibition of proinflammatory transcription factor activity (Madrigal-Matute et al., 2010). In addition, it has been confirmed that 17-DMAG could protect renal adenocarcinoma (ACHN) cells from an H2O2-mediated oxidative stress (Harrison et al., 2008).
The Akt signaling pathway has been shown to mediate cell proliferation, growth, and survival in many systems (Hu et al., 2016). In cardiomyocytes, activation of Akt can be beneficial to not only cell survival but also cell function following H/R injury both in vitro and in vivo (Pachori et al., 2007; Parvin et al., 2014). Nuclear factor erythroid 2-like 2 (Nrf2), the central transcription factor, regulates multiple lines of cellular antioxidant systems, which is important in the protection of cells against oxidative stress (Hu et al., 2016). Heme oxygenase-1 (HO-1) is a phase II defense enzyme that possesses potent antioxidative ability. The overexpression of HO-1 in the brain of mice confers neuroprotective effects against H/R (Hu et al., 2016). Interestingly, 17-DMAG has been demonstrated to markedly induce HO-1 expression after LPS initiation in rats (Wang et al., 2016b). Recent studies have paid close attention to the Akt/Nrf2/HO-1 pathway that plays a key role in oxidative stress and neuronal survival (Yan et al., 2015). In this study, we investigated whether 17-DMAG could activate the Akt/Nrf2/HO-1 pathway.
A previous study has shown that H/R treatment can be used to induce cell injury in hippocampal neurons (Tamatani et al., 2000). Therefore, in our study, the effects of 17-DMAG were explored on H/R-induced cell viability, cytotoxicity, and apoptosis in hippocampal neurons (HT22). Moreover, we evaluated the protective effects of 17-DMAG on the expression of antioxidant genes and its possible mechanism.
Materials and Methods
Cell culture
HT22 murine hippocampal neuronal cells (ATCC, Rockville, MD) were cultured in DMEM supplemented with 10% (v/v) FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified 5% (v/v) CO2 incubator at 37°C. One day before the experiments, these cells were seeded in six-well culture dishes (106 per well).
Cellular models of H/R
For the H/R experiments, hypoxic conditions were created by incubating the cells in an anaerobic chamber equilibrated with 2.5% O2, 5% CO2, and 92.5% N2 at 37°C for 24 h. The cells were then reoxygenated under normoxic conditions in a humidified atmosphere (95% air/5% CO2) at 37°C for 24 h. Normoxic control cells were incubated at 37°C under 95% air/5% CO2. The H/R cells were treated with 17-DMAG or vehicle (water). In the 17-DMAG experiments, the cells were pretreated for 1 h with 17-DMAG (100, 250, 500 nM) and then subjected to hypoxia and reoxygenation.
Quantification of cell damage
Cell damage was determined by measuring cell viability and the release of lactate dehydrogenase (LDH) into the cell culture medium. The release of LDH was quantified using the CytoTox-ONE™ Homogenous Membrane Integrity Assay (Promega Corp., Madison, WI) according to the manufacturer's instructions. Cell viability was determined using an MTT assay as described elsewhere (Choi et al., 2010). After removing the medium, phosphate-buffered saline (PBS) and MTT were added in 96-well plates to prepare 0.5 mg/mL MTT. The microplate was incubated at 37°C for an additional 3 h. At the end of the incubation period, the medium containing MTT was removed and 200 mL of dimethyl sulfoxide was added into each well. The plate was shaken on the microplate shaker to dissolve the blue MTT-formazan. Absorbances were read at a wavelength of 570 nm on a microplate reader (Bio-Rad, Hercules, CA).
Measurement of cell apoptosis
Apoptosis of cells was examined by double staining with Annexin V-FITC and PI. HT22 cells (1 × 105 cells/well) were seeded into six-well culture plates and incubated in normoxic condition or H/R condition with or without 17-DMAG. After treatment, cells were resuspended in PBS and 500 μL binding buffer containing 10 μL Annexin V-FITC stock and 10 μL PI (20 μg/mL). After incubation for 15 min at room temperature in the dark, the samples were analyzed immediately by flow cytometry. The Annexin V+/PI− cells were considered apoptotic cells and the fluorescence intensity was analyzed by Cellquest™ software (Becton-Dickinson, CA).
ROS detection
ROS production was measured by flow cytometry using DCFH-DA staining. DCFH-DA was cleaved intracellularly by nonspecific esterases and transformed to highly fluorescent 2′,7′-dichlorofluorescein (DCF) in the presence of ROS. Briefly, after treatment, the cells were incubated with DCFH-DA for 30 min at 37°C in dark. After incubation, cells were washed twice with PBS. Fluorescence intensity was measured by the microplate reader (Molecular Devices, CA) at an excitation wavelength of 495 nm and an emission wavelength of 520 nm. The level of ROS was expressed as percentages of values against the control.
Preparation of nuclear and cytoplasmic extractions
Nuclear and cytoplasmic extractions were performed using the Nuclear and Cytoplasmic Extraction kit (Sigma-Aldrich, St. Louis, MO) following manufacturer's instructions. The cells were harvested and washed with 1 mL buffer A (1.5 mM MgCl2, 18 mM KCl, 10 mM HEPES, pH 7.9) for 5 min at 600 g and then resuspended in buffer A and 0.1% NP 40, left for 10 min on ice to lyse the cells, and centrifuged for 3 min at 600 g. The supernatant was saved as cytosolic extract. The nuclear pellet was washed in 1 mL buffer A for 3 min at 4000 g, resuspended in 30 μL buffer C (25% glycerol, 0.42 M NaCl, 20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA), rotated at 4°C, and then centrifuged for 20 min at 14,000 g. The supernatant was saved as nucleus extract.
Western blotting
Equal amounts of cell protein were resolved on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels (Sigma-Aldrich) and transferred onto nitrocellulose membrane (Schleicher and Schuell, Keene, NH). After blocking with 5% nonfat milk, the blots were probed using primary antibodies against cleaved HSP90 (1:500), HSP70 (1:500), Hop (1:1000), caspase-3 (1:1000), Bax (1:1000), Bcl-2 (1:1000), p-Akt (1:1000), Akt (1:1000), nuclear Nrf2 (1:1000), MnSOD (1:500), CAT (1:500), GPx (1:300), lamin-B (1:1000), and β-actin (1:1000). Detection was by incubation with goat anti-mouse IgG (1:5000; Sigma-Aldrich) followed by enhanced chemiluminescence (Amersham Pharmacia, NJ). The intensity of the bands was measured using Image Quant software.
Real-time reverse transcription–polymerase chain reaction
Total RNA was prepared using the Trizol reagent (Invitrogen, Carlsbad, CA) and the optical absorbance ratio at 260 nm/280 nm was measured to determine the content. Then, the RNA was reverse transcribed using a High-Capacity cDNA Archive kit (Applied Biosystems) into cDNA with random hexamer primers. The reverse transcription products were used for amplification using the SYBR Green PCR amplification reagent (Qiagen, Valencia, CA). Quantitative real-time reverse transcription–polymerase chain reaction analysis was performed with cDNA as the template to amplify MnSOD, CAT, and GPx mRNAs with specific primers. The mRNAs were normalized to β-actin mRNA with the comparative Ct method. The primers for qPCR are listed in Table 1.
CAT, catalase; GPx, glutathione peroxidase; MnSOD, manganese superoxide dismutase.
Statistical analysis
All data were expressed as the mean ± SD of results derived from three independent experiments performed in triplicate. Statistical analysis was performed by the Student's t-test and analysis of variance. A difference was accepted as significant if p < 0.05.
Results
17-DMAG protects HT22 cells from H/R-induced cytotoxicity
The expression levels of heart shock proteins HSP90, HSP70, and cochaperone protein Hop in H/R-induced cells were detected by Western blot assay. The results showed that the level of HSP90 was reduced by 17-DMAG in HT22 cells. Moreover, the expression of HSP90, HSP70, and Hop were downregulated in the H/R-induced cells, which were upregulated after 17-DMAG treatment (Fig. 1A, B). The effects of 17-DMAG on H/R-induced cell injury in HT22 cells were evaluated by MTT and LDH assays. The results showed that 17-DMAG had slight cell toxicity to the normoxic control group. However, the H/R treatment markedly reduced cell viability and induced cytotoxicity. Treatment with 17-DMAG led to a dose-dependent recovery of cell viability (Fig. 1C, D). These results suggest that 17-DMAG protects HT22 cells from H/R-induced cytotoxicity in a dose-dependent manner.
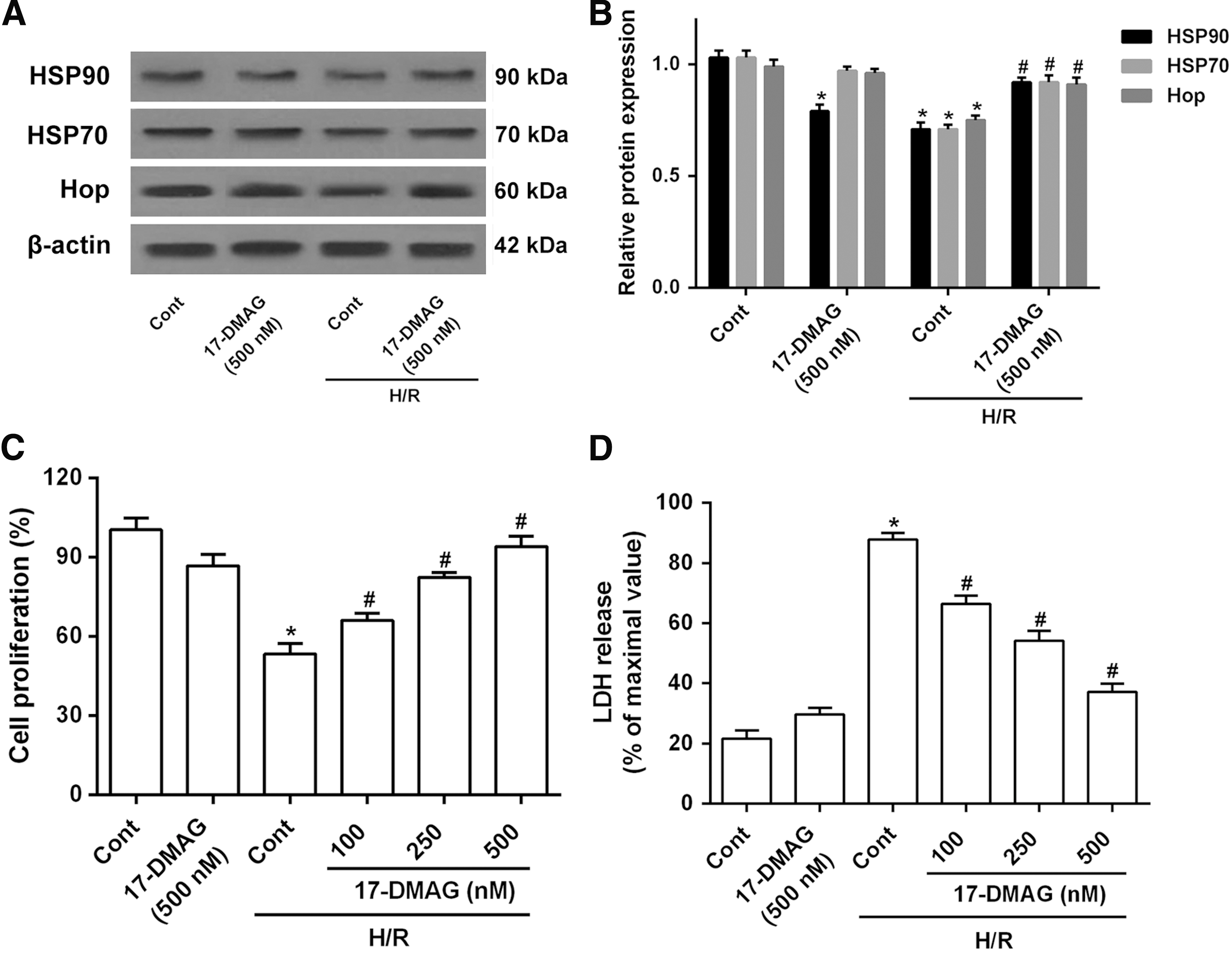
17-DMAG protects HT22 cells from H/R-induced cytotoxicity.
17-DMAG protects HT22 cells from H/R-induced apoptosis
Flow cytometry analysis showed that both early apoptosis and late apoptosis were upregulated in H/R-treated HT22 cells compared to control cells. After treatment with 17-DMAG (100, 250, and 500 nM), the cell apoptosis rate was decreased in H/R-treated HT22 cells (Fig. 2A). Moreover, the expression of caspase-3 and Bax/Bcl-2 was significantly increased in H/R-treated HT22 cells, but dramatically reduced in 17-DMAG-treated HT22 cells compared with the control cells (Fig. 2B). These data indicated that 17-DMAG inhibits cell apoptosis induced by H/R in HT22 cells in vitro.
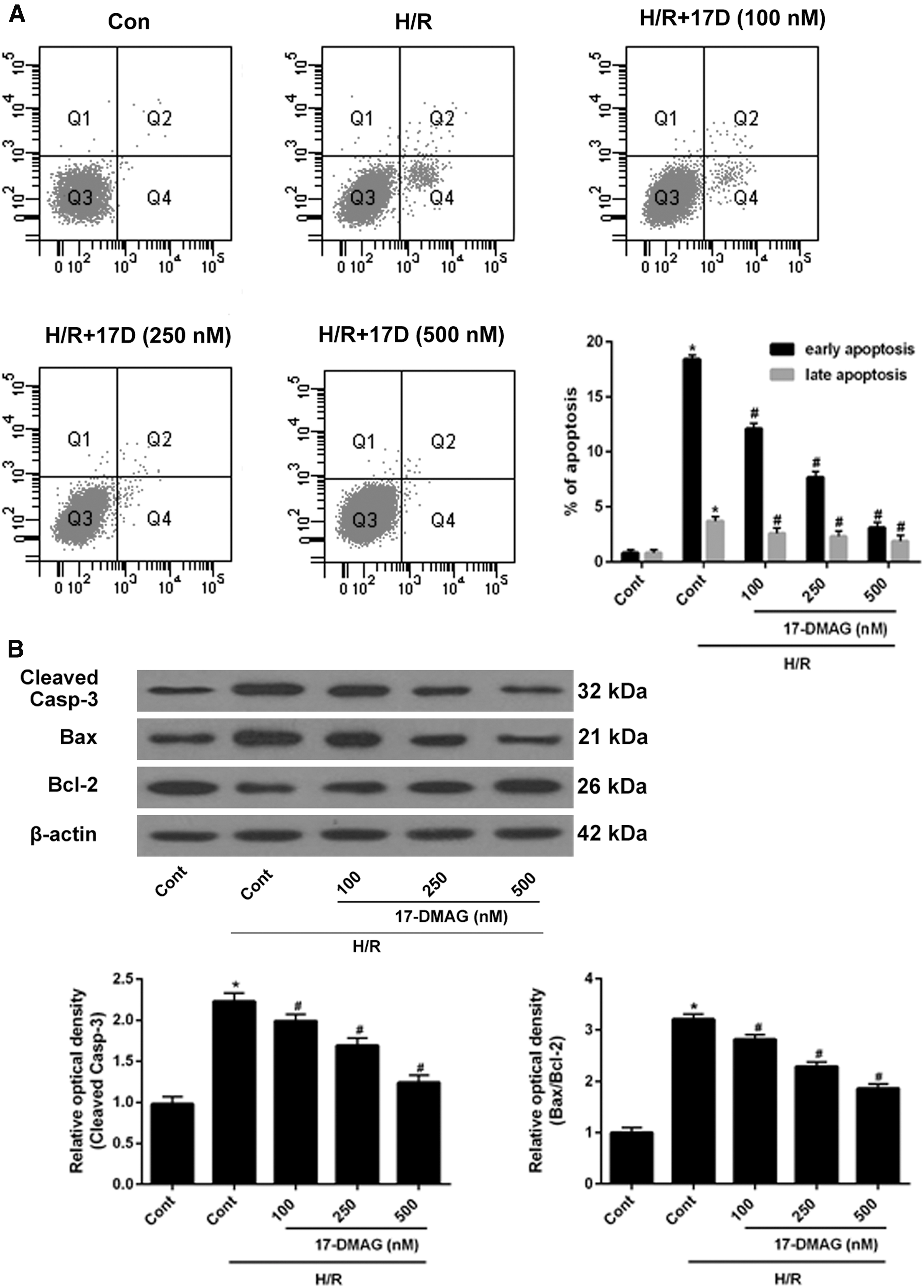
17-DMAG protects HT22 cells from H/R-induced apoptosis. HT22 cells were treated with 17-DMAG of different concentrations for 24 h,
17-DMAG protects HT22 cells from H/R-induced oxidative stress
As an initial experiment toward determining the role of 17-DMAG in the regulation of cellular antioxidant enzymes and attenuation of oxidative stress induced by H/R, HT22 cells were treated with different concentrations of 17-DMAG after being subjected to H/R. 17-DMAG markedly attenuated H/R-induced elevation of ROS content (Fig. 3A), indicating that 17-DMAG resisted H/R-induced oxidative stress in HT22 cells. In addition, H/R reduced the levels of endogenous MnSOD, CAT, and GPx protein. However, these protein expression levels were upregulated by 17-DMAG in a dose-dependent manner (Fig. 3B). Consistently, the mRNA levels of MnSOD, CAT, and GPx were also induced by 17-DMAG. Overall, these findings demonstrated that 17-DMAG induced the expression of antioxidant enzymes and attenuated H/R-induced oxidative stress in HT22 cells.
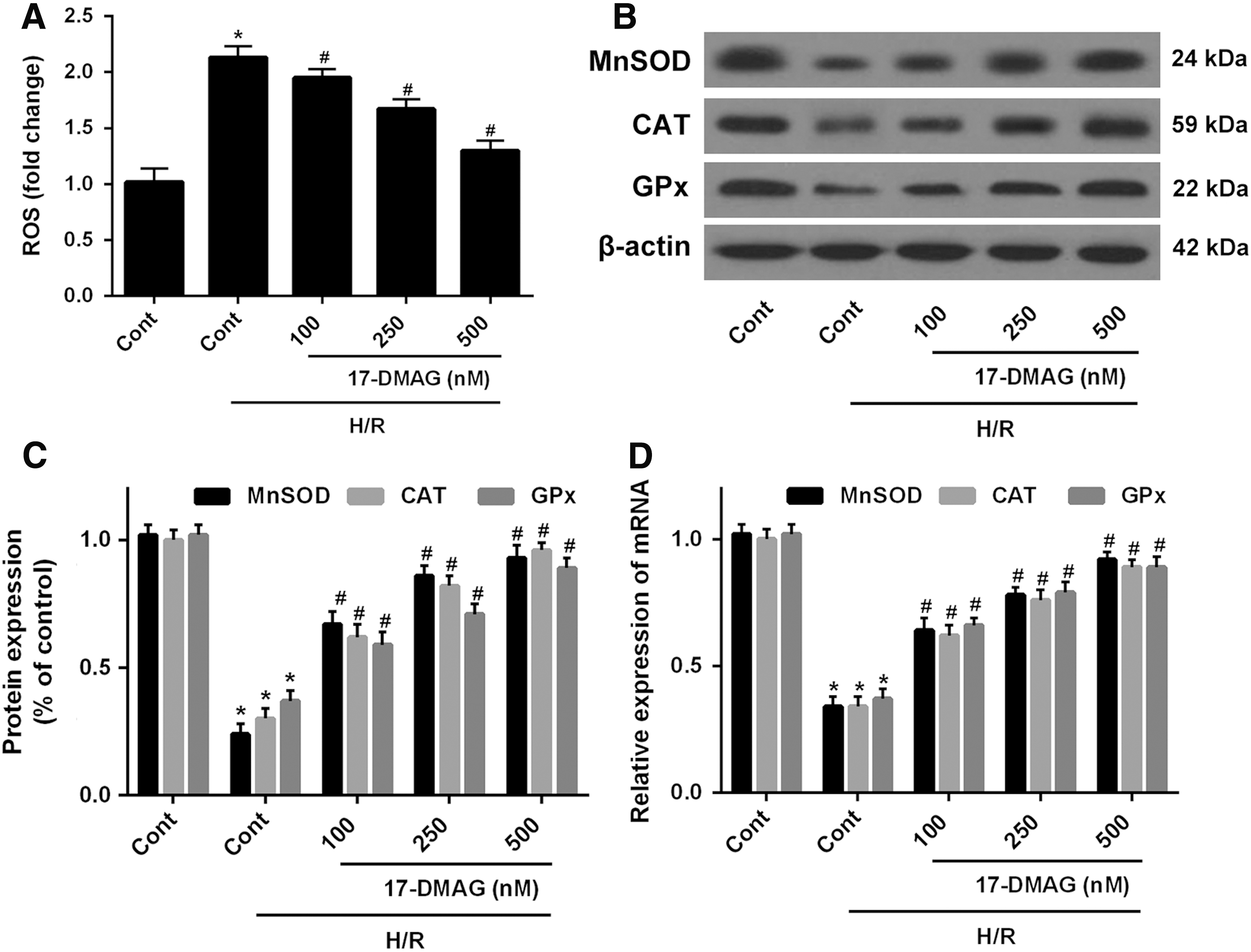
17-DMAG protects HT22 cells from H/R-induced oxidative stress. HT22 cells were treated with 17-DMAG of different concentrations for 24 h,
17-DMAG protects against H/R-induced cells injury in HT22 cells by activating Akt/Nrf2/HO-1 pathway
To investigate the mechanism of 17-DMAG on H/R-induced oxidative stress, the expression of oxidative stress-related proteins nuclear Nrf2 and HO-1 was measured. The expression of p-Akt, nuclear Nrf2, and HO-1 was markedly reduced by H/R, but treatment with 17-DMAG (500 nM) significantly increased the expression of p-Akt, nuclear Nrf2, and HO-1 protein expression in H/R-treated HT22 cells, whereas, Akt inhibitor LY294002 markedly abolished the effects of 17-DMAG on p-Akt expression and blocked the expression of nuclear Nrf2 and HO-1 (Fig. 4A). In addition, the results showed that LY294002 abolished the effects of 17-DMAG on H/R-induced cell viability and apoptosis (Fig. 4B, C). Moreover, the effects of 17-DMAG on the decrease of antioxidant gene levels induced by H/R were also abolished by LY294002 (Fig. 4D). These findings demonstrated that 17-DMAG might induce expression of antioxidant genes by activating the Akt/Nrf2/HO-1 pathway.
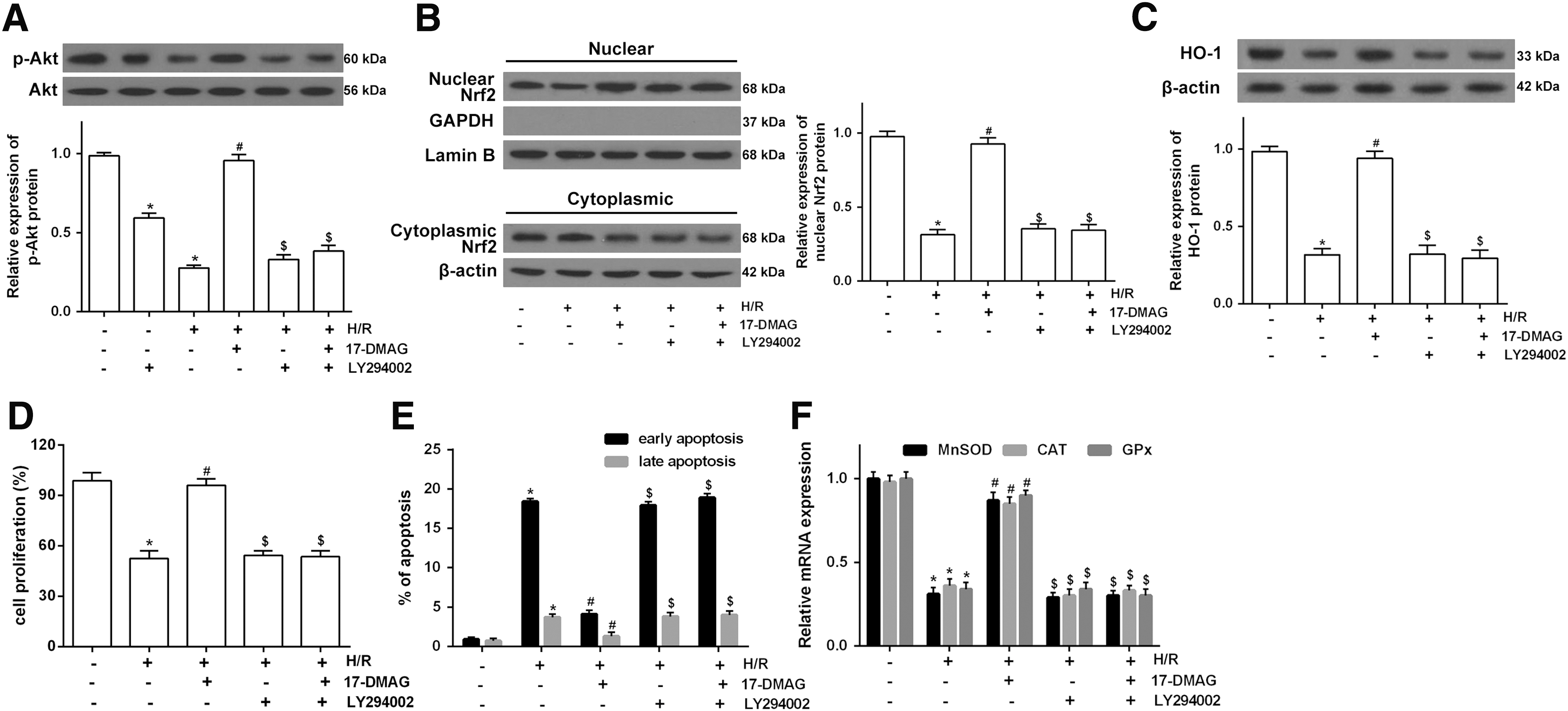
17-DMAG induces expression of antioxidant gene by activating the Akt/Nrf2/HO-1 pathway.
Discussion
CVD ranks as the top three health risks, especially cerebral ischemia characterized with the damage of hippocampal neurons. H/R injury, as a primary event in the pathogenesis of neurodegeneration, is characterized with neuronal cell death (Yang et al., 2014). In this study, we evaluated the neuroprotective effects of 17-DMAG on H/R-induced cell injury and the underlying mechanisms in HT22 mouse hippocampal cells. 17-DMAG strongly inhibits neuroblastoma cell growth by targeting both anaplastic lymphoma kinase and N-myc (Yi et al., 2014). It has been reported that 17-DMAG inhibits necroptotic cell death by the downregulation of receptor-interacting proteins (Park et al., 2015). In this study, cell viability and cytotoxicity were assessed using the MTT and LDH assays, and the results suggested that 17-DMAG protects HT22 cells from H/R-induced cytotoxicity. In addition, the expression of HSP90, HSP70, and Hop was downregulated in H/R-induced HT22 cells, which was upregulated by 17-DMAG. This is consistent with the previous study that indicated heat shock protein 90 binding agents (17-DMAG) upregulate the expression of heat shock proteins and alleviate ischemia–reperfusion-induced cell injury (Harrison et al., 2008).
Apoptosis plays an important role in the loss of hippocampal neurons in a variety of pathologies, including H/R injury (Wei et al., 2015). The expression of Bcl-2 family members, such as Bcl-2 and Bax, plays key roles in the cell apoptosis process. Bax is a proapoptotic protein, whereas Bcl-2 is an antiapoptotic protein. It has been noted that the ratio of Bcl-2 to Bax acts as an essential element in determining the threshold of apoptosis (Hu et al., 2016). In addition, caspase-3 has been found to be activated by the apoptotic pathway and then processed into activated fragments such as cleaved caspase-3, which are considered an index of apoptosis (Salvesen, 2002). In this study, the expression of caspase-3 and Bax/Bcl-2 was significantly increased in H/R-treated HT22 cells, but dramatically reduced with different concentrations of 17-DMAG in H/R-treated HT22 cells.
The hippocampus contains the highest densities of excitatory amino acid receptors in the mammalian brain, which renders hippocampal neuron more susceptible to oxidative stress and neurodegenerative complications (Liu et al., 2013). Accumulation of ROS causes DNA oxidation and lipid peroxidation leading to cellular apoptosis or death (Yan et al., 2015). Hypoxic or reoxygenated mitochondria can produce excess ROS. The generation of ROS and its consequent cellular responses are the hallmark of H/R-mediated hippocampal neuron apoptosis (Wei et al., 2015). It has been confirmed that 17-DMAG protected renal ACHN cells from an H2O2-mediated oxidative stress. In addition, 17-DMAG diminished ROS production and further interfered with oxidative stress in vascular smooth muscle cells. The reduced levels of antioxidant enzymes, including MnSOD, CAT, and GPx, are characteristic of H/R-induced oxidative stress (Yan et al., 2015). Furthermore, this study showed that H/R suppressed the induction of these enzymes and further augmented the oxidative stress-induced injury, which was diminished by 17-DMAG treatment. However, the mechanisms involved in the induction of these antioxidative enzymes by 17-DMAG have remained elusive, and therefore, this study focused on clarifying the underlying mechanisms.
Recent studies have paid close attention to the Akt/Nrf2/HO-1 pathway that plays a key role in oxidative stress and neuronal survival. Nrf2 is the primary transcription factor that mediates transcriptional expression of numerous antioxidant genes (including MnSOD, CAT, and GPx) by binding to the ARE element present in the promoters of these genes (Li et al., 2015). Emerging studies have demonstrated that HO-1 has a crucial role in the protection of cells against oxidative stress (Abass et al., 2016). Actually, Nrf2 is a key regulator of HO-1 expression in cells (Sun et al., 2016). Moreover, the PI3K/Akt pathway has been demonstrated to be associated with activated Nrf2/HO-1 (Kim et al., 2010). Of note, 17-DMAG has been reported to be a stabilizer and positive regulator of HO-1, aiding antioxidant gene activation (Madrigal-Matute et al., 2012; Wang et al., 2016b). On this basis, it was hypothesized that activation of the Akt/Nrf2/HO-1 pathway may be responsible for the induction of antioxidative enzymes by 17-DMAG and contribute to the protective functions of 17-DMAG against H/R-induced oxidative stress in HT22 cells. In this study, Akt phosphorylation was significantly enhanced in the HT22 cells treated with 17-DMAG compared with that observed in the H/R group. 17-DMAG enhanced the expression of nuclear Nrf2 and HO-1, but LY294002 partly blocked the increase in Nrf2 translocation to the nucleus and the upregulation of HO-1 expression. Furthermore, the effects of 17-DMAG on H/R-induced cell proliferation and apoptosis were markedly negated by LY294002. Our results revealed that 17-DMAG conferred an effect on the activation of the Akt/Nrf2/HO-1 pathway in H/R-treated hippocampal neuron, which is a potential mechanism for enhancing the antiapoptotic and antioxidant effects.
Conclusion
In conclusion, we found that 17-DMAG significantly attenuates H/R-induced hippocampal neuron injury in vitro by exerting antioxidant and antiapoptotic effects through the Akt/Nrf2/HO-1 signaling pathway. These observations demonstrated the importance of the 17-DMAG/Nrf2 axis in the cellular response to oxidative stress-induced cell death, suggesting that this axis may be a potential therapeutic target for CVDs associated with oxidative stress such as cerebral ischemia reperfusion injury.
Footnotes
Disclosure Statement
No competing financial interests exist.