
Abstract
Alpha-synuclein (α-syn) is a highly conserved protein encoded by the SNCA gene and is expressed uniquely in neurons of both the central and peripheral nervous systems (CNS and PNS). α-Syn is known to cause sporadic and familial forms of Parkinson's disease (PD). However, the role for neuronal expression of α-syn in the first place remains unknown. We review and discuss recently published work that suggests a novel role for α-syn expression in neurons as a restriction factor that inhibits virus transmission from the PNS to the CNS. The potential new role for α-syn expression as a virus inhibitor may provide new approaches to understand the pathogenesis of PD and provide novel approaches to prevent and treat this common neurodegenerative disease.
Introduction
A
Pathogenesis of PD and Viral Infections
There is a long history of links between viral infections and PD (Harris et al., 2012; Bu et al., 2015; Boyd et al., 2016). Although it is unclear that any specific viral infection causes PD, the epidemiologic links continue to suggest that viral exposure over time may increase risk for PD. Whether this is due to a direct effect of the virus on dopaminergic neurons or a result of the immune response is not clear. During acute West Nile virus (WNV) encephalitis, acute viral infection of the substantia nigra pars compacta occurs in some patients, resulting in clinical Parkinsonian symptoms including cog-wheel rigidity, bradykinesia, and wide-based gait (Sejvar et al., 2003; Bode et al., 2006; Sejvar, 2007; Petersen et al., 2013). These patients may recover from acute loss of dopaminergic signaling and improve over time. Thus, the direct effect of viral-induced injury to dopaminergic neurons is not clearly linked to the long-term process of α-syn-induced injury of dopaminergic neurons seen in sporadic PD.
Based on these observations, we proposed a novel hypothesis. If α-syn functioned to inhibit viral infections in neurons as a restriction factor, then repeated virus-induced changes in α-syn expression in neurons over time may place genetically susceptible individuals at risk for the development of PD. In support of this novel hypothesis, PD patients develop prodromal symptoms commonly characterized by changes in smell and/or gastrointestinal (GI) symptoms before clinical onset of PD (Kalia and Lang, 2015; Mahlknecht et al., 2016). The olfactory bulb and the GI neurons are common sites of viral neuroinvasion into the CNS. The current theory in the PD field states that an environmental trigger results in post-translational modifications in α-syn in peripheral GI neurons and/or the olfactory bulb promoting fibrilization, and then α-syn fibrils spread to the CNS in a prion-like process (Dehay et al., 2016; Rey et al., 2016a, 2016b). Based on our clinical observations that neuroinvasive viruses from diverse families and genera (enteroviruses, flaviviruses, herpesviruses, and alphaviruses) commonly cause peripheral infections but rarely cause CNS disease, and that complex nervous systems are continuously exposed to potential viral infections in the GI tract and exposed olfactory neurons, we hypothesized that α-syn expression should inhibit virus neuroinvasion from peripheral sites to the CNS.
Alpha-synuclein expression inhibits virus infection of the CNS
We examined the role of α-syn expression in viral encephalitis using α-syn knockout mice and our mouse model of WNV encephalitis (Beatman et al., 2015). Using a murine model of WNV encephalitis, mice expressing α-syn as syngeneic wildtype, heterozygous, or knockout for α-syn were peripherally inoculated with WNV. To our surprise, α-syn knockout mice exhibited markedly increased virus-induced weight loss, mortality, and caspase 3-dependent neuronal injury. These phenotypes were associated with dramatically increased viral growth in the brain compared with heterozygous or wildtype mice (Beatman et al., 2015). Our initial work implied that α-syn expression inhibited viral infection of the CNS after peripheral inoculation of WNV. To test our hypothesis that α-syn inhibited transport of viral infections from the PNS to the CNS, we used an alphavirus, Venezuelan equine encephalitis virus TC83, that is attenuated and is not neuroinvasive in black6 mice. After peripheral inoculation of TC83, we found very high viral loads of TC83 in the brains of α-syn knockout mice but found no evidence of virus infection in the brains of mice expressing α-syn as heterozygotes (Beatman et al., 2015). These data again supported our hypothesis that α-syn expression inhibits neuroinvasion from the PNS to the CNS.
In support of these initial conclusions, we have observed no difference in viral growth comparing wild-type and α-syn knockout mice in the brain after intracerebral inoculation of WNV or after inoculation of virus directly on brain slice cultures, thus bypassing peripheral routes of infection. In our previous work, we found a small reduction in WNV growth after infection of α-syn knockout primary cortical neurons compared with wild-type neurons (Beatman et al., 2015). We found that this small reduction was likely related to accelerated neuronal death in α-syn knockout primary cortical neurons. This is in stark contrast to a mean increase in viral titer of 104.5 plaque forming units (pfu) in the brains of α-syn knockout mice after peripheral inoculation in the footpad of 1000 pfu of WNV.
One possible explanation for the potential specificity of α-syn function for PNS to CNS transport is the intracellular localization of α-syn after viral infection. We have previously shown that α-syn increases localization to Rab1+ membranes in the exocytosis pathway after viral infection (Beatman et al., 2015). We also found that α-syn colocalizes with WNV envelope protein (Fig. 1). Taken together, these data imply that α-syn localizes to membranes in the exocytosis pathway of neurons to prevent neuron-to-neuron spread during infection.
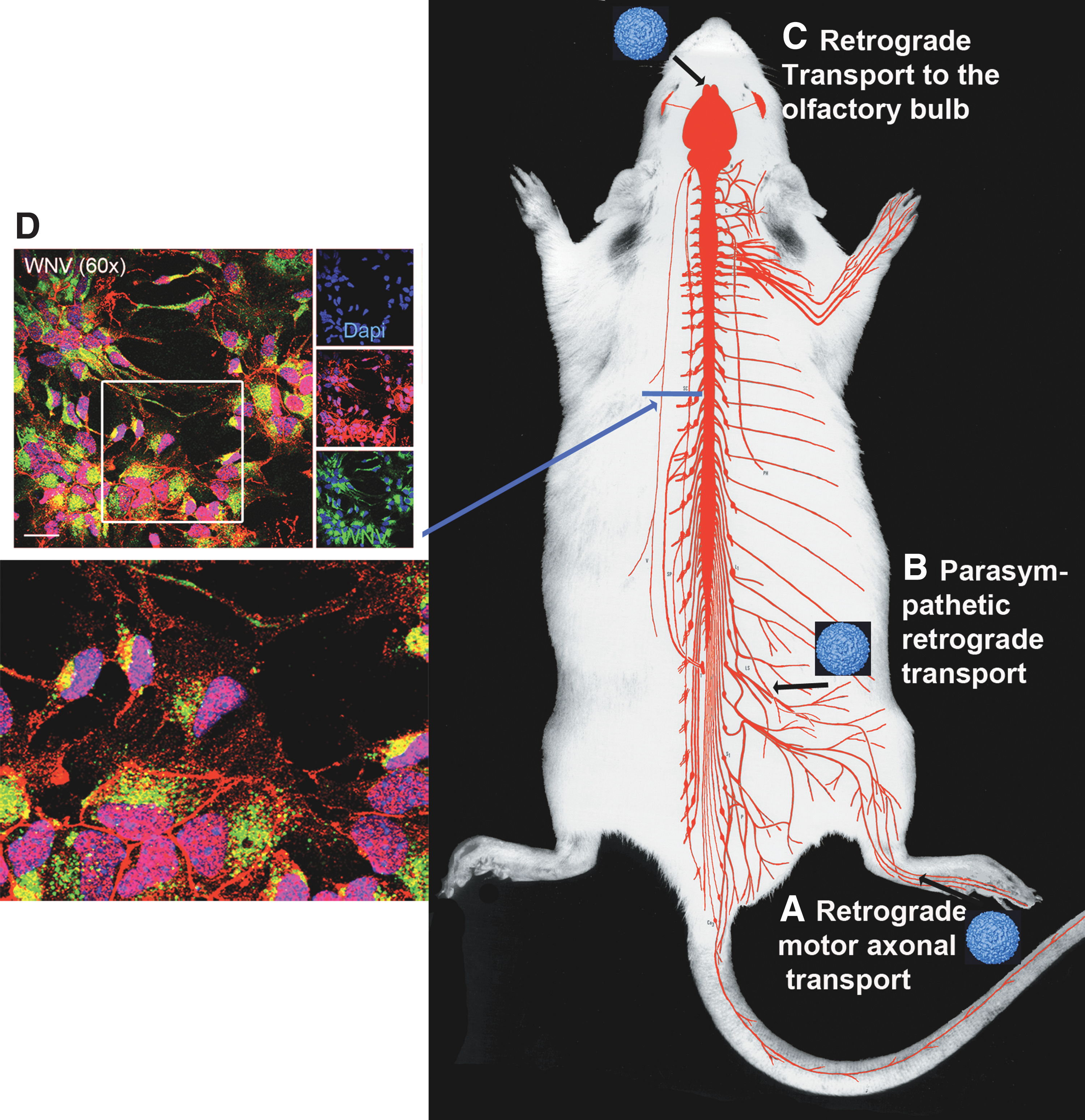
Working model for alpha-synuclein-induced inhibition of viral neuroinvasion. Potential routes of virus transneuronal spread from the peripheral nervous system to the central nervous system (black arrows) include retrograde spread from
Conclusion
Although much is known about the etiology of PD with regard to α-syn, little is known about the native function of α-syn under normal physiologic conditions or what specific event(s) trigger α-syn to form fibrils and initiate the pathogenesis of PD. Our initial data suggest a stunning new paradigm in the fields of neurovirology and PD research: α-syn functions as a novel viral restriction factor in complex nervous systems. Our findings further support the hypothesis that viral restriction is most likely achieved by inhibiting viral exocytosis pathways, thus preventing neuron-to-neuron spread of virus from the PNS to CNS (Fig. 1). Current work on the pathogenesis of PD states that an environmental trigger initiates post-translational changes in α-syn, resulting in fibrilization of α-syn and subsequent prion-like spread from the PNS (e.g., GI and olfactory neurons) into the CNS, resulting in dopaminergic neuron loss and PD progression. However, little is known about the environmental triggers responsible for the initial post-translational changes in α-syn. Additional studies evaluating the role of virus-induced post-translational changes of α-syn in the PNS are definitively needed to further define the role of viral infections in post-translational modification to α-syn. Future studies of these interactions will provide evidence of a potential role of recurrent virus exposure in the PNS as a potential cause for α-syn fibrilization and prion-like spread.
Footnotes
Acknowledgments
We would like to thank Dr. Patrik Brundin at the Van Andel Institute for many insightful discussions and collaborative approaches during the development of our work. The Beckham laboratory is currently funded by University of Colorado School of Medicine institutional funds, the University of Colorado Center for Neuroscience (CNS) pilot funding, and the Department of Neurology at the University of Colorado School of Medicine.
Disclosure Statement
No competing financial interests exist.