
Abstract
The fibroblast growth factor (FGF) 16 gene (Fgf-16) is preferentially expressed by neonatal cardiomyocytes after birth, with levels increasing into adulthood. Null mice and isolated heart studies suggest a role for FGF-16 in cardiac maintenance and survival, including increased resistance to doxorubicin (DOX)-induced injury. However, the effect of DOX on endogenous FGF-16 synthesis and specifically regulation of cardiac Fgf-16 expression has not been reported. Here we assess the effect of DOX on FGF-16 RNA levels and stability as well as promoter activity and use sequence analysis, knockdown, and overexpression to investigate the role of cardiac transcription factor(s) implicated in the response. Endogenous FGF-16 RNA levels were reduced >70% in 8-week-old rats treated with 15 mg DOX/kg for 6 h. This was modeled in neonatal rat cardiomyocyte cultures, where an equivalent decrease was also seen within 6 h of 1 μM DOX treatment. Six kilobases of mouse Fgf-16 upstream flanking and promoter DNA was also assessed for DOX responsiveness in transfected cardiomyocytes. A decrease in FGF-16 promoter activity was seen with only 747 base pairs containing the Fgf-16 TATA box that includes a putative and highly conserved binding site for the cardiac transcription factor Csx/Nkx2.5. There was also no effect of DOX on FGF-16 RNA stability, consistent with transcriptional control. Levels and binding of Csx/Nkx2.5 to the FGF-16 promoter were reduced with DOX treatment. Knockdown of Csx/Nkx2.5 specifically decreased endogenous FGF-16 RNA and protein levels, whereas Csx/Nkx2.5 overexpression stimulated levels, and increased resistance to the rapid DOX-induced depletion of FGF-16. These observations indicate that Fgf-16 expression is directly regulated by Csx/Nkx2.5 in neonatal cardiomyocytes, and a negative effect of DOX on Csx/Nkx2.5 and, thus, endogenous FGF-16 synthesis may contribute indirectly to its cardiotoxic effects. Targeting FGF-16 levels could, however, offer increased resistance to cardiac injury.
Introduction
H
There is, however, an apparent switch from limited endocardial and epicardial synthesis to predominantly myocardial production by the heart in the perinatal period, which reaches its highest levels in the myocytes of the adult heart from where it was cloned (Miyake et al., 1998; Lavine et al., 2005; Lu et al., 2008b). No other member of the FGF gene family is reported to share this temporal and spatial pattern of expression. Thus, taken together with its structural conservation between species, this indicates cardiac-specific control and an important role for FGF-16 and, by extension, a need to understand its regulation (Wang et al., 2015).
Evidence from null mice and isolated heart studies suggests that FGF-16 plays a role in survival of cardiomyocytes and maintenance of the myocardium. Specifically, in an angiotensin II-induced model of cardiac injury, both cardiac hypertrophy and fibrosis were increased in FGF-16 null mice (Matsumoto et al., 2013). In addition, FGF-16 was able to partially rescue the impaired regenerative capability and heart function in neonatal GATA4 null mice by inhibiting hypertrophy and apoptosis, reducing scar tissue formation as well as stimulating replication of cardiomyocytes (Yu et al., 2016).
An important role for FGF-16 is also suggested by the higher incidence of cardiac-associated abnormalities in human families with nonsense mutations of FGF-16 (Laurell et al., 2014). Finally, FGF-16 offered increased resistance to acute injury by the anticancer drug doxorubicin (DOX) in an isolated mouse heart model (Sontag et al., 2013). DOX is widely used in chemotherapy, but is associated with an increased and cumulative risk of cardiac damage, thereby limiting its usefulness/efficiency (Octavia et al., 2012). While endogenous FGF-16 is expected to offer some resistance to cardiomyocytes from DOX-related damage, there is no report on how Fgf-16 expression is regulated in response to DOX treatment.
Multiple transcription factors have been implicated in the expression and regulation of Fgf-16 in cardiomyocytes. A link between GATA4 and Fgf-16 expression was indicated in the neonatal mouse heart, and a GATA4 DNA element was detected in the second intron of mouse Fgf-16 (Yu et al., 2016). Sequence analysis and/or in vitro studies with the Fgf-16 promoter have also suggested roles for stress-related nuclear factor-kappaB (NF-κB), as well as the cardiac transcription factors myocyte enhancer factor (MEF) 2 and the helix motif homeodomain protein Csx/Nkx2.5 (Sofronescu et al., 2008; 2010; Wang et al., 2015).
In the case of Csx/Nkx2.5, an equivalent putative binding site is located within human, rat, and mouse Fgf-16 TATA sequences (Wang et al., 2015). In addition to conservation of their putative DNA elements within Fgf-16 sequences, both Csx/Nkx2.5 and GATA4 contribute to cardiomyocyte development (Schlesinger et al., 2011), can regulate each other (Searcy et al., 1998), and participate in a common complex in vitro and in vivo (Durocher et al., 1997). Csx/Nkx2.5 is also required for homeostasis and survival of cardiomyocytes in the adult heart and is a known target of DOX (Toko et al., 2002). Importantly, in terms of the current study, Csx/Nkx2.5 and GATA4 offer protection to cardiomyocytes against DOX-induced cell death (Toko et al., 2002; Aries et al., 2004).
In this study, we have assessed the effect of DOX on Fgf-16 expression in rat hearts and neonatal rat cardiomyocyte cultures. Fgf-16 is activated preferentially in these cells of the newborn rat myocardium (Miyake et al., 1998; Lu et al., 2008b), and they have been used extensively to study protection from injury, including DOX-induced cytotoxic damage (Wang et al., 2013; Sun et al., 2014). Our data provide evidence of a role for Csx/Nkx2.5 in the expression of Fgf-16, and the negative regulation by DOX observed.
Materials and Methods
DOX treatment and neonatal rat cardiomyocyte cultures
All procedures involving animals, their tissues, and cells conform to the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th Edition. Revised 2011), and were approved by the local animal Protocol Management and Review Committee. Eight-week-old Sprague–Dawley rats were treated with 15 mg DOX/kg body weight or saline vehicle by intraperitoneal (i.p.) injection (Zordoky et al., 2010; Alimoradi et al., 2012). Rats were euthanized after 6 h and hearts harvested for isolation of RNA. For cultures, 1-day-old Sprague–Dawley rats were euthanized by decapitation, and ventricular myocytes isolated by enzymatic digestion and fractionation on a Percoll gradient. Cells were plated in the presence of serum for 24 h and then cultured in defined medium as previously described (Wang et al., 2013). Cells were then treated with 1 μM DOX (Sigma-Aldrich, Oakville, Canada) for 2, 6, and/or 24 h.
RNA isolation, and real-time reverse transcriptase–polymerase chain reaction
RNA extraction and qPCR using specific primers (Table 1) were done as described (Sofronescu et al., 2010). Minus reverse transcriptase (RT) reactions were performed as controls for the presence of genomic DNA. RNA levels in each sample (relative quantification) were normalized to rat RNA polymerase (pol) II expression. Tests were normally run in duplicate on three independent samples.
Protein extraction and immunoblotting
Neonatal rat cardiomyocyte nuclear extract (NE) was isolated for the detection of Csx/Nkx2.5 using the EpiSeeker Nuclear Extraction Kit (Abcam, Inc., ON, Canada) according to the manufacturer's instructions. FGF-16 was also isolated from culture medium using heparin sepharose beads (GE Healthcare, Toronto, ON, Canada, CL-6B), prepared by adding an equal volume of 0.5 M NaCl/10 mM Tris. Twenty-five μL of bead solution was added to each 1.5 mL of medium and mixed overnight at 4°C. Beads were washed with 200 μL 0.5 M NaCl/10 mM Tris before adding 30 μL of sodium dodecyl sulfate (SDS) protein sample buffer to collect the protein.
Proteins (5 μg of nuclear protein and all extractions from the culture medium) were separated by 10% and 15% SDS sodium dodecyl sulfate–polyacrylamide gel electrophoresis (PAGE), respectively. Proteins were transferred to polyvinylidene fluoride (PVDF) membrane and immunoblotted with either anti-Csx/Nkx2.5 (cat # sc-8697; Santa Cruz, Dallas, TX) or anti-FGF-16 antibodies (Abcam; #ab170515). Proteins were visualized using horseradish peroxidase-conjugated anti-immunoglobulin G (IgG) secondary antibody and ECL plus immunoblotting detection reagents (Thermo Fisher Scientific, Inc., Nepean, ON, Canada). Ponceau Red staining of the membrane was used to compare the relative loading of total protein extracted by heparin from the medium of test neonatal rat cardiomyocyte cultures. For nuclear protein assessed by immunoblotting, lamin B (cat # sc-6217; Santa Cruz) was used as a loading control.
Transient transfection and the luciferase reporter gene assay
Generation of hybrid Luc genes driven by different lengths of mouse (m) Fgf-16 promoter (p) sequences was described previously (Sofronescu et al., 2008). This includes −5771/−12, −3772/−12, −1267/−12, and −759/−12 FGF-16p.Luc. The nucleotide designation for each promoter fragment is given relative to the adenine residue (A), identified as nucleotide +1, of the methionine (ATG) translation start site of mFGF-16. Neonatal rat cardiomyocytes were seeded at 3.5 × 105 cells/well (12-well culture plates) for 1 day before gene transfer using the TransIT-2020 transfection reagent (Mirus, Madison, WI). A total of 2 μg of test plasmid, or empty vector as a negative control (pcDNA 3.1), was used per well. Cells were also cotransfected with the Renilla-luciferase (R-Luc) gene (20 ng) as a control for DNA uptake (Sofronescu et al., 2010). After 18 h, cultures were refed with defined medium before DOX treatment for 2, 6, and 24 h. Luciferase activity was assessed in a luminometer (Lumat LB 9507) using a dual luciferase reporter assay (Promega, Dual-Luciferase® Reporter Assay System, E1960).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed essentially as previously described (Sofronescu et al., 2008), using neonatal rat cardiomyocyte NE and a 32P-labeled oligonucleotide (5′-ACGTATATAAAGTGCCATTT-3′) probe containing TATA1 sequences and a putative Csx/Nkx2.5-binding site. These sequences (nucleotides −312/−292) are identical in both the rat and mouse Fgf-16 promoter regions. For treatment with antibodies, Csx/Nkx2.5 antibody (Ab) and mouse IgG were added and preincubated on ice for 10 min before adding radiolabeled probes. The DNA-protein complexes were resolved in nondenaturing 5% (w/v) polyacrylamide gels and visualized by autoradiography.
Chromatin immunoprecipitation-qPCR assay
Immunoprecipitation (ChIP)-qPCR assay (FactorPath™) was performed by Active Motif (Carlsbad, CA). ChIP reactions were done using 20 μg of neonatal rat cardiomyocyte chromatin and 20 μL of Csx/Nkx2.5 and NF-κB (p65) antibodies (Santa Cruz; cat # sc-8697 and sc-109). Primers for specific regions of interest as well as an untranslated region on chromosome 17 (Untr17) as a negative control were used for qPCR. Atrial natriuretic factor gene (Nppa-310) and nuclear factor of kappa light polypeptide gene enhancer in B cell inhibitor, alpha (Nfkbia +1002) sequences, contain known binding sites for Csx/Nkx2.5 and NF-κB (p65) binding, respectively, (van Essen et al., 2009; Warren et al., 2011), and were thus assessed as positive controls using specific primers (Table 1).
Small interference RNA-mediated knockdown
Cellular transfection to knockdown Csx/Nkx2.5 expression was performed using HiPerFect Transfection Reagent according to the manufacturer's protocol in neonatal rat cardiomyocytes (Qiagen, cat # 301705). Csx/Nkx2.5 siRNA-1/2 (FlexiTube siRNA, cat # SI01920338, Rn_Nkx2-5/1, target sequence 5′-CGCCTACGGCTACAACGCCTA-3′ and SI01920352, Rn_Nkx2-5/2, and target sequence 5′-GACCCTCGGGCGGATAAGAAA-3′) was purchased from Qiagen. The knockdown was done in 12-well culture plates at a cell density of 3.5 × 105 cells/well using 50 nM siRNA and control siRNA (Qiagen; cat # SI03650318).
Adenovirus concentration qPCR titration and adenovirus-mediated gene delivery
Adenoviral DNA was purified using the NucleoSpin Virus kit (MACHEREY-NAGEL, Bethlehem, PA; cat#740977.10). Briefly, 150 μL of rat cardiomyocyte lysate harvested after adenoviral transfection was treated with deoxyribonuclease I (DNase I) to remove any residual host cell or plasmid DNA carried over from cell packaging, and viral DNA isolated after Proteinase K treatment. Serial dilution combined with qPCR was done to determine the threshold cycle for each dilution using the Adeno-X qPCR Titration Kit (Clontech, Mountain View, CA; cat# 632252). DNA copy number and multiplicity of infection (MOI) were then calculated from a standard curve generated by plotting the Ct values of the serial diluted (six orders of magnitude) Adeno-X DNA control template.
Premade Csx/Nkx2.5 (cat # 00454A, Human-Csx/Nkx2.5-AdV), CMV Null (cat # 000047A, CMV-AdV), and HA-GATA4 (a gift from Dr. Mona Nemer, University of Ottawa, Rat-GATA4-AdV) overexpression adenoviruses, were amplified in HEK293 cells according to the manufacturer's protocol (Applied Biological Materials, Inc., Richmond, BC, Canada). Adenovirus transfection (overexpression) was performed in 12-well multiple culture plates at a cell density of 3.5 × 105 cells/well at a MOI of 10 and/or 50 for 1 h. After washing, cells were refed with medium and incubated for 48 h before treatment with 1 μM DOX for 12 h before isolation and assessment of RNA by qPCR.
Statistical analyses
Paired t-tests were applied for single comparisons, and one-way analysis of variance (ANOVA) with a post hoc Tukey's test as well as two-way ANOVA with a post hoc Bonferroni test were used for multiple (treatments and time) group analyses. Mean values were considered significantly different if p < 0.05; */#/‡ p < 0.05, **/##/‡ ‡ p < 0.01, and ***/###/‡ ‡ ‡ p < 0.001. Unless stated otherwise, all studies were done in triplicate.
Results
FGF-16 RNA levels are rapidly decreased in response to DOX treatment
To assess the effect of DOX on endogenous FGF-16 RNA levels, rats were treated with or without a single i.p. injection of DOX (15 mg/kg body weight) for 6 h. This dose is reported to induce acute cardiomyopathy in the rat (Zordoky et al., 2010; Alimoradi et al., 2012). Heart RNA was isolated and FGF-16 transcript levels were determined by qPCR. A significant >70% decrease in FGF-16 RNA levels with DOX relative to saline injection was detected (Fig. 1A). To model this effect for further study, neonatal rat cardiomyocyte cultures were treated with 1 μM DOX for 2, 6, and 24 h, and total RNA was isolated and assessed by qPCR. There was a significant >70% decrease in FGF-16 RNA levels at 2 h that was reduced further (>90%) at 6 and 24 h (Fig. 1B).

DOX decreases FGF-16 RNA levels in rat heart and neonatal cardiomyocytes. FGF-16 RNA levels were assessed by qPCR in:
To assess a possible effect of DOX on FGF-16 RNA stability, cardiomyocytes were treated with 2 μg/mL of the transcription inhibitor actinomycin D (ActD) for 2.5 h before 1 μM DOX treatment, and assessment of FGF-16 RNA levels by qPCR. As expected, FGF-16 RNA levels were decreased in response to ActD treatment. The calculated “slope” indicates a FGF-16 RNA half-life of ∼1.75 h (Fig. 1C). There was, however, no significant effect of DOX treatment on the pattern of FGF-16 RNA degradation (Fig. 1C).
FGF-16 is released by cardiomyocytes in culture (Lu et al., 2008b). Thus, the effect of 1 μM DOX on FGF-16 protein levels in the medium was also assessed by heparin extraction, gel electrophoresis, and immunoblotting using specific antibodies to FGF-16 (Fig. 1D). Multiple low-mobility (>50 kDa) bands were observed, as well as a band of ∼26.5 kDa that is consistent with glycosylated and released FGF-16 (Lu et al., 2008b). 1 A decrease in the intensity of this band was seen after treatment with 1 μM DOX for 24 h (arrow, Fig. 1D).
DOX treatment decreases FGF-16 promoter activity
Hybrid firefly luciferase (Luc) genes driven by 5.8–0.8 kb of mouse FGF-16 promoter sequences were used to assess the effect of DOX on promoter (p) activity in transfected neonatal rat cardiomyocytes. A promoterless (-p) Luc gene was also assessed, and all Luc genes were cotransfected with a Renilla (R)-Luc gene as a control for DNA uptake. Transfections were done for 18 h, and then cells were fed with defined medium containing 1 μM DOX for 2, 6, and 24 h, before luciferase activity (Luc/R-Luc.) was measured. All FGF-16p.Luc genes generated activity significantly greater than -p.Luc (Fig. 2A), and no significant effect of DOX on -p.Luc was observed within 24 h (Fig. 2B). Similarly, no effect of DOX on FGF-16 p.Luc gene expression was observed at 2 h. However, luciferase activity was decreased significantly after 6 h DOX treatment with all four FGF-16 p.Luc genes, and a further decrease was seen at 24 h compared with controls (Fig. 2C–F).

DOX treatment decreases hybrid Fgf-16 promoter/firefly luciferase (Luc) reporter gene activity.
The reduction in FGF-16 RNA levels is associated with a DOX-induced decrease in Csx/Nkx2.5 levels and binding at the FGF-16 proximal promoter region
The 747 bp mouse FGF-16 promoter region containing a single TATA box (TATA1) was sufficient to confer responsiveness to DOX treatment (Wang et al., 2015). A comparison reveals a high degree of similarity between mouse, rat, and human sequences in this region and by extension conservation of putative transcription factor-binding sites (Wang et al., 2015). This includes a site for Csx/Nkx2.5 at nucleotide position −305/−299 (5′-TAAAGTG-3′), relative to the ATG start codon for rat and mouse FGF-16, where A is designated as nucleotide position +1. This possible Csx/Nkx2.5 DNA element overlaps with a TATA box sequence (underlined) in the human, rat, and mouse FGF-16 promoter (5′-
ChIP assay was done to assess the effect of DOX treatment on Csx/Nkx2.5 and NF-κB (p65) association with the FGF-16 TATA1 box region in situ (Fig. 3A, B). Chromatin from neonatal rat cardiomyocytes treated without or with 1 μM DOX for 2 and 6 h was crosslinked, fragmented (∼500 bp), and immunoprecipitated with specific antibodies to Csx/Nkx2.5 and NF-κB (p65). Specific PCR primers spanning nucleotides −396/−315 of Fgf-16 were used to detect fragments associated with Csx/Nkx2.5 and NF-κB (p65) binding in the proximal promoter (TATA1 at nucleotide −307) region by qPCR (Table 1). An untranslated (Untr17) region was used as the negative control. In addition, primers for Nppa-310 and Nfkbia +1002 gene sequences containing known binding sites for Csx/Nkx2.5 and NF-κB (p65), respectively, were also used as positive controls.

The reduction in FGF-16 RNA levels is associated with a DOX-induced decrease in Csx/Nkx2.5 association with the FGF-16 promoter. Association of Csx/Nkx2.5 and NF-κB (p65) with FGF-16 promoter sequences in neonatal rat cardiomyocytes treated with or without 1 μM DOX was assessed by ChIP in situ, using specific antibodies to
Significant Csx/Nkx2.5 binding above background (Untr17) levels was seen with both Nppa-310 and FGF-16 sequences at both 2 and 6 h (Fig. 3A). A significant decrease in Csx/Nkx2.5 binding to Nppa-310 and FGF-16 sequences was observed at 2 h with DOX treatment, and this was reduced further at 6 h (Fig. 3A). By contrast, no significant binding of NF-κB (p65) to Fgf-16 sequences was detected in either control or DOX-treated samples at 2 and 6 h (Fig. 3B). Significant NF-κB (p65) binding to Nfkbia +1002 sequences was seen in the control at 6 h, but was decreased with DOX treatment (Fig. 3B). No significant change was observed at 2 h (Fig. 3B).
To assess whether Csx/Nkx2.5 levels are affected under conditions where DOX decreases FGF-16 gene expression, neonatal rat cardiomyocytes were treated with 1 μM DOX for 2, 6, and/or 24 h. RNA was then assessed by qPCR, and effects on protein levels and DNA binding were assessed by protein immunoblotting and EMSA, respectively, using nuclear proteins and specific antibodies to Csx/Nkx2.5 (Fig. 3C–F). Csx/Nkx2.5 RNA levels were significantly decreased at 2 h, and further decreases were seen until 24 h (Fig. 3C). A corresponding decrease in Csx/Nkx2.5 protein is also suggested in response to DOX treatment (Fig. 3D).
For EMSA, a radiolabeled 20 bp oligonucleotide corresponding to nucleotides −311/−292 and containing the FGF-16 TATA1 box and Csx/Nkx2.5 DNA element was incubated with neonatal rat cardiomyocyte NE. A pattern of low-mobility complexes was detected. The lowest mobility complex was competed with a specific antibody to Csx/Nkx2.5, but not mouse immunoglobulins (Fig. 3E). To assess the effect of DOX on this binding, the same 20 bp oligonucleotide was combined with NE from neonatal rat cardiomyocytes treated without or with 1 μM DOX for 0.5, 2, 6, and 24 h. Although no decrease was seen at 30 min, a reduction in low mobility complexes was observed with 2 h DOX treatment and at all subsequent time points relative to untreated controls (Fig. 3F).
Fgf-16 expression is responsive to Csx/Nkx2.5 overexpression and knockdown
The effect of Csx/Nkx2.5 levels on endogenous FGF-16 RNA was investigated in neonatal rat cardiomyocytes. For Csx/Nkx2.5 overexpression, cells were transfected with adenovirus (10 MOI) expressing human Csx/Nkx2.5 (Csx/Nkx2.5-AdV), rat GATA4 (GATA-AdV), or a null adenovirus under the control of the cytomegalovirus (CMV-Null-AdV) promoter for 1 h. The culture medium was changed and cells were maintained for 48 h. Increased levels of Csx/Nkx2.5 and GATA4 gene expression were confirmed (Fig. 4A, B). Overexpression of Csx/Nkx2.5 significantly and specifically increased FGF-16 RNA levels as, by comparison, FGF-2 transcripts were decreased (Fig. 4C, D). There was also no effect of GATA4 overexpression alone or when combined with Csx/Nkx2.5 overexpression as, in the case of the latter, FGF-16 RNA levels were not significantly different compared with Csx/Nkx2.5 overexpression only (Fig. 4C).
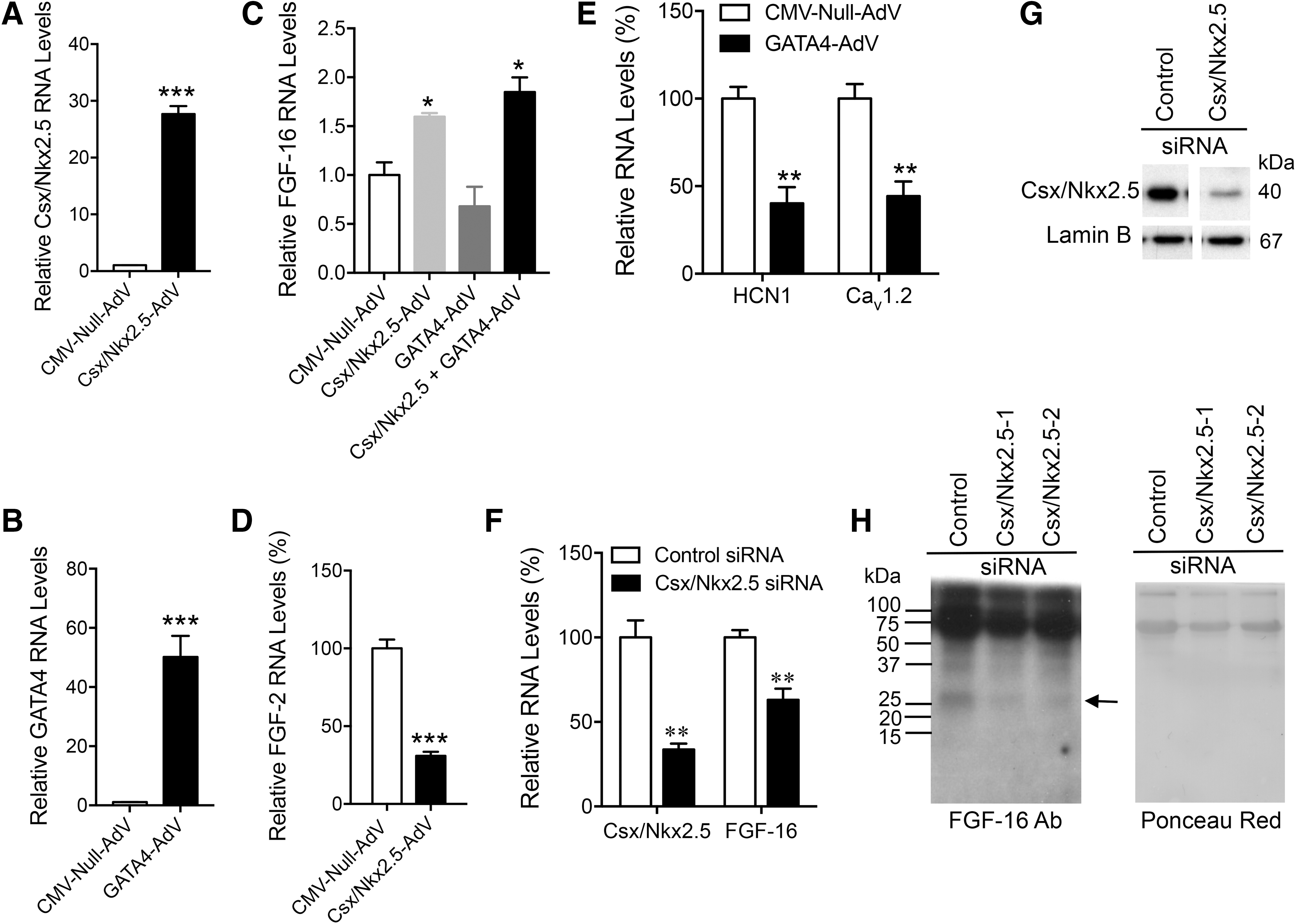
Csx/Nkx2.5 levels regulate FGF-16 gene expression. Neonatal rat cardiomyocytes were transfected with 10 MOI CMV-Null AdV, Csx/Nkx2.5-AdV, and GATA4-AdV for 48 h.
By contrast, overexpression of GATA4 did significantly downregulate two known GATA4 targets as reported by others (Wang et al., 2007), specifically, potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 1 (HCN1) and calcium channel, voltage-dependent, L-type, alpha 1C subunit (Cav 1.2) RNA levels (Fig. 4E). For knockdown, cardiomyocytes were transfected with 50 nM of rat Nkx2.5 siRNA (Csx/Nkx2.5 siRNA-1/2) or a scrambled control siRNA for 72 h. A significant decrease in Csx/Nkx2.5 RNA and protein levels was detected with Nkx2.5 siRNA relative to control siRNA (Fig. 4F, G). A corresponding and significant 36.9% decrease in FGF-16 transcripts was seen specifically with Csx/Nkx2.5 siRNA knockdown (Fig. 4F), and this was reflected in reduced detection of FGF-16 protein in the culture medium (Fig. 4H).
Overexpression of Csx/Nkx2.5 limits the negative effect of DOX on FGF-16 RNA levels
Rapid depletion of FGF-16 transcripts by DOX is associated with decreased Csx/Nkx2.5 levels and evidence of binding to the FGF-16 promoter. Thus, the ability of increased Csx/Nkx2.5 expression to resist the downregulation of endogenous FGF-16 levels in response to DOX treatment was assessed. Neonatal rat cardiomyocytes were transfected with 10 MOI adenovirus-expressing Csx/Nkx2.5 and CMV-Null AdV for 1 h. The culture medium was changed, and after 48 h, cells were treated with 1 μM-DOX for 12 h, before isolation and assessment of FGF-16 RNA by qPCR and protein by immunoblotting.
Csx/Nkx2.5 AdV overexpression alone induced FGF-16 RNA and protein levels significantly (Fig. 5). In addition, a significant and greater than 80% decrease in FGF-16 RNA levels was seen with DOX treatment of cells transfected with CMV-Null AdV. By contrast, the DOX-induced decrease in FGF-16 transcripts in neonatal rat cardiomyocytes transfected with Csx/Nkx2.5 AdV recovered significantly by 20% (Fig. 5A). Csx/Nkx2.5 AdV transfection before DOX treatment also limited a DOX-induced reduction in FGF-16 protein detected in the culture medium (Fig. 5B).

Overexpression of Csx/Nkx2.5 limits the negative effect of DOX on FGF-16 RNA levels. Neonatal rat cardiomyocytes were transfected with 10 MOI CMV-Null AdV or Csx/Nkx2.5-AdV for 48 h cells before treatment with 1 μM DOX for 12 h.
Discussion
There is evidence that FGF-16 is an endogenous maintenance and survival factor for postnatal cardiomyocytes, and that it may increase protection from cardiac injury, including against acute DOX-related damage (Matsumoto et al., 2013; Sontag et al., 2013; Wang et al., 2015; Yu et al., 2016). However, in this study, we detected a relatively rapid and significant decrease in FGF-16 transcript levels using a single i.p. dose of DOX that is associated with acute cardiomyopathy (Zordoky et al., 2010; Alimoradi et al., 2012). This response was modeled in neonatal rat cardiomyocytes and assessed using a combination of RNA, protein, gene transfer, as well as overexpression and knockdown studies.
The FGF-16 gene was shown to be specifically regulated by the cardiac homeodomain transcription factor Csx/Nkx2.5, which is negatively targeted by DOX. Increasing and decreasing Csx/Nkx2.5 levels resulted in corresponding elevated and reduced FGF-16 levels, respectively. Csx/Nkx2.5 is known to regulate a number of cardiac genes, including cardiac α-actin, atrial natriuretic peptide (ANP), cardiac ankyrin repeat protein, and myocardin (Ueyama et al., 2003; Akazawa and Komuro, 2005). In addition, Csx/Nkx2.5 levels and association with the Fgf-16 promoter were negatively targeted by acute DOX treatment, resulting in a rapid decrease in FGF-16 synthesis (within 2 h) and secretion detected at 24 h. DOX is known to adversely affect cardiac transcription factors, including Csx/Nkx2.5, as well as GATA4 and MEF2C (Kobayashi et al., 2010; Zheng et al., 2013).
This is consistent with negative effects of DOX on the expression of multiple cardiac-specific genes (Boucek et al., 1999). This, in turn, may help to explain the cardiotoxicity of DOX, and specifically the damage done to heart structure and contractile function compared with other tissues at an early stage. Finally, overexpression of Csx/Nkx2.5 was able to significantly rescue FGF-16 expression in the presence of DOX. These data suggest that endogenous levels of FGF-16 may offer limited benefit against DOX-induced cardiac injury, unless amplified or supplemented.
In addition to regulating Fgf-16 expression in neonatal cardiomyocytes, as indicated above, our data also suggest a role for the cardiac-specific factor Csx/Nkx2.5 in the negative response of FGF-16 to DOX treatment. Activity of a truncated mouse 747 bp Fgf-16 promoter region that contains a Csx/Nkx2.5 site within the TATA1 region, but not GATA4 or MEF2 DNA elements, was reduced significantly in response to DOX treatment. Furthermore, DOX decreased Csx/Nkx2.5 availability and binding to Fgf-16 promoter sequences containing TATA1, which is conserved in the human Fgf-16 promoter (Toko et al., 2002; Zheng et al., 2013; Wang et al., 2015). Csx/Nkx2.5 is downregulated by DOX, and transgenic mice overexpressing a dominant negative form of Csx/Nkx2.5, as well as knockdown of Csx/Nkx2.5 in neonatal rat cardiomyocytes, results in less resistance to DOX-induced cardiomyocyte apoptosis (Toko et al., 2002; Zheng et al., 2013).
Complementary to these observations, increasing Csx/Nkx2.5 availability was shown to protect cardiomyocytes from DOX-induced apoptotic cell death (Toko et al., 2002; Zheng et al., 2013). This protection may be related, at least in part, to a corresponding increase in FGF-16 availability, given FGF-16 production was stimulated by Csx/Nkx2.5 overexpression, and cardioprotective activity of FGF-16 was suggested in isolated heart studies (Sontag et al., 2013). By extension, however, the selective and rapid decrease in FGF-16 production in response to DOX treatment is expected to reduce resistance to cardiac injury.
Our data do not exclude a role for additional transcription factors in the DOX-induced decrease in Fgf-16 expression, as Csx/Nkx2.5 overexpression was only able to partially limit the negative effect of DOX on FGF-16 RNA levels. GATA4 is a candidate, as selective knockdown of GATA4 expression in the neonatal mouse heart was associated with a decrease in FGF-16 production, suggesting that Fgf-16 might be a direct target for GATA4 (Yu et al., 2016). Furthermore, GATA4 is rapidly depleted by DOX in neonatal rat cardiomyocytes (Aries et al., 2004; Yu et al., 2016). However, unlike the stimulation seen with Csx/Nkx2.5, no increase in FGF-16 RNA levels was seen with adenoviral overexpression (10 MOI) of GATA4, and even when the MOI was increased to 50 (data not shown), although control channel gene RNAs were affected as reported (Wang et al., 2007).
In addition, the 747 bp proximal mouse Fgf-16 promoter region was responsive to DOX, but does not contain a consensus GATA DNA element, although a GATA4-binding site has been described in the second intron of Fgf-16 (Yu et al., 2016). It is possible that a DNA-binding site is not required for GATA in the Fgf-16 promoter region as GATA4 can also act as a coactivator of Csx/Nkx2.5 (Jiang et al., 1999). However, we did not obtain support for this possibility based on the results of combined adenoviral overexpression of GATA4 and Csx/Nkx2.5 in our cardiomyocyte cultures. Specifically, there was no significant difference in the effect of GATA4 and Csx/Nkx2.5 versus Csx/Nkx2.5 overexpression alone on FGF-16 RNA levels.
Although not cardiac-specific, the proximal Fgf-16 promoter region does contain a NF-κB DNA element that is conserved between species and binds NF-κB (p50 and p65) in situ (Sofronescu et al., 2010). However, while the decrease in FGF-16 RNA levels with DOX treatment of neonatal cardiomyocytes corresponded with a decrease in Csx/Nkx2.5 binding at the promoter in situ, no change in NF-κB (p65) association was detected. While these data do not rule out any role for GATA4 or NF-κB, they are consistent with Csx/Nkx2.5 as a significant target to explain the negative effect of DOX on Fgf-16 promoter activity.
Evidence largely from mouse studies suggests that FGF-16 plays an important role in development and maintenance of a healthy myocardium (Lavine et al., 2005; Hotta et al., 2008; Lu et al., 2008a; Matsumoto et al., 2013; Sontag et al., 2013; Wang et al., 2015; Yu et al., 2016). As a consequence, endogenous FGF-16 appears to limit the effects of cardiac injury and/or remodeling, including hypertrophy and fibrosis (Matsumoto et al., 2013; Yu et al., 2016). By contrast, while treatment with exogenous FGF-16 was able to increase resistance to acute DOX-induced damage in an isolated mouse heart (Sontag et al., 2013), endogenous FGF-16 is unlikely to offer more than limited cardioprotection given the negative effects on new synthesis.
In addition to the relatively short (1.75 h) half-life determined for FGF-16 RNA, DOX treatment resulted in a rapid and negative effect on Csx/Nkx2.5 levels and binding to the Fgf-16 promoter in cardiomyocytes. Thus, rapid depletion may compromise cardiomyocyte viability and decrease resistance to injury at a very early stage of DOX exposure, given the evidence for FGF-16 as a cardiac maintenance and survival factor. Cardiac complications can happen at any time during DOX treatment or even years after the last chemotherapy session (Octavia et al., 2012). In some cases injury may occur immediately after a single dose or course of DOX therapy, resulting clinically in transient electrophysiological abnormalities within 24 h, a pericarditis, myocarditis syndrome, or acute left ventricular failure (Shakir and Rasul, 2009; Lipshultz et al., 2013).
In summary, we show for the first time that the Fgf-16 promoter binds and is regulated by the cardiac transcription factor Csx/Nkx2.5. Furthermore, FGF-16 synthesis is rapidly and negatively targeted by DOX in the heart in vivo and cardiomyocyte cultures in vitro. This is related, at least in part, to a decrease in availability and binding of Csx/Nkx2.5 to the Fgf-16 promoter, and specifically TATA-related sequences that are conserved between human and murine species. Thus, supplementing or maintaining FGF-16 levels by targeting components of the synthetic pathway may help reduce the risk of DOX-induced damage in cardiomyocytes.
Footnotes
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research. J.W. is the recipient of a Research Manitoba Doctoral Studentship.
Disclosure Statement
No competing financial interests exist.