
Abstract
Zika virus (ZIKV) is a mosquito-borne flavivirus. It was first isolated from Uganda in 1947 and has become an emergent event since 2007. However, because of the inconsistency of alignment methods, the evolution of ZIKV remains poorly understood. In this study, we first use the complete protein and an alignment-free method to build a phylogenetic tree of 87 Zika strains in which Asian, East African, and West African lineages are characterized. We also use the NS5 protein to construct the genetic relationship among 44 Zika strains. For the first time, these strains are divided into two clades: African 1 and African 2. This result suggests that ZIKV originates from Africa, then spread to Asia, Pacific islands, and throughout the Americas. We also perform the phylogeny analysis for 53 viruses in genus Flavivirus to which ZIKV belongs using complete proteins. Our conclusion is consistent with the classification by the hosts and transmission vectors.
Introduction
Z
Until 2015, the strain of ZIKV emerged in the city of Natal in Brazil (Zanluca et al., 2015). Soon after, autochthonous transmission of ZIKV took place throughout other countries in the Americas. After the outbreak in Brazil, a significant increase of microcephaly in newborns was documented by the Brazilian Ministry of Health. Some authors propose that the microcephaly is possibly caused by ZIKV (Broutet et al., 2016). Humans infected with ZIKV present a dengue-like syndrome related to mild fever, headache, cutaneous rashes, retroorbital pain, and conjunctivitis (Faye et al., 2014). This clinical characteristic could easily be mistaken for dengue or chikungunya fevers, which are two common diseases associated with public health.
ZIKV is a positive-sense single-stranded RNA virus, with an ∼11 kb genome. The genome contains a single open reading frame (ORF) with flanking 5′ and 3′ untranslated regions. The ORF encodes a polyprotein that is cleaved into three structural proteins: the capsid, precursor of membrane, and envelope (E), and seven nonstructural proteins: NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. Among them, the NS5 protein is the largest viral protein, of which C-terminus is related to RNA polymerase activity and the N-terminus is involved in RNA capping.
As far as we know, the phylogenetic trees of ZIKV were all constructed by multiple sequence alignment algorithms and based on genomic sequences. However, these trees were inconsistent with one another. For example, the recovered evolutionary relationship presented two cases: Asian and African lineages; Asian and two African lineages. Based on the complete genomes, the two different cases of ZIKV phylogeny could be obtained (Enfissi et al., 2016; Faria et al., 2016; Lanciotti et al., 2016). Using the E and NS5 genes, although some studies generated three lineages, the topology of trees was different (Faye et al., 2014; Shen et al., 2016). The phylogeny derived from partial genes may be two lineages as well (Zhu et al., 2016). Moreover, the proposed hypothesis that ZIKV is originated from Africa has not been verified (Gong et al., 2016). The protein sequences of ZIKV are directly involved in a variety of biological processes and more conserved than the gene sequences. To fully understand the origin and diversity of this emergent agent, it is necessary to perform phylogeny analysis by alignment-free methods with protein sequences.
Over the past decade, the number of alignment-free methods for inferring phylogeny of organisms increased sharply. Many of them are variants of k-word methods, which only take advantage of the occurrence of words of length k (Dai et al., 2008; Wu et al., 2009). The recently proposed natural vector method includes information from both positions and counts of amino acids (Deng et al., 2011). As shown in the previous work (Yu et al., 2013), the natural vector method can achieve more accurate results than alignment-based methods for phylogeny analysis. The method is also fast in computational speed compared with alignment-based ones.
The investigation for other flaviviruses in the genus Flavivirus, to which ZIKV belongs, has been limited due to the lack of sequenced genomes and their inability to infect vertebrates. In recent years yet, some of the viruses caused several global epidemics or became localized public health concern. These viruses are small, enveloped, single-stranded and positive-sense ssRNA viruses, which infect various hosts such as birds, mammals and insects. Their genomes are ∼11 kb in length and have a similar structure to ZIKV (Billoir et al., 2000; Moureau et al., 2015). Due to the wide diversity among these flaviviruses, knowledge of their genetic relationships is limited and mainly comes from alignment-based methods. In this study, the evolutionary relationships of ZIKV strains and other flaviviruses are constructed by the natural vector methods with protein sequences. We also verify the hypothesis for the Africa origin of ZIKV.
Materials and Methods
Source of datasets
There is only one reference genome from ZIKV in NCBI and many ZIKV strains have short partial genes. Some complete genomes of ZIKV strains also show difference in length. Since the complete coding region of ZIKV encodes a long protein (polyprotein), which is subsequently processed into 10 proteins, including the major NS5 protein, the polyprotein sequences of 87 ZIKV strains were retrieved from NCBI on 23 June 2016. The accession numbers, countries where the viruses are isolated, the collection dates, and length of polyproteins are listed in Supplementary Table S1; Supplementary Data are available online at
The proteins translated from the NS5 gene were obtained from the 87 complete proteins. Among them 43 protein sequences are identical to others and then only the remaining 44 protein sequences are kept in subsequent analysis. The polyprotein sequences of 53 referenced genomes in the Flavivirus genus were extracted from NCBI as well, and their accession numbers are listed in Supplementary Table S2.
Natural vector
The recently proposed natural vector method is a powerful tool for virus classification and phylogeny (Deng et al., 2011). This method is alignment free and needs no parameters. By this approach, each protein sequence is mapped into a 60-dimensional numeric vector, which has been proven to perform well to capture information in viral sequences (Yu et al., 2013; Huang et al., 2014; Li et al., 2016). We review the method as follows. Let
(1) Let
(2) Let
(3) Let
For ambiguous amino acids, the letter J indicates it may be L or I; B for D or N; Z for Q or E; and X for all possible 20 types of amino acids. Thus, for
Thus, the 60-dimensional natural vector of a protein sequence S is defined as
The Euclidean distance is a true mathematical distance that satisfies the triangle inequality, that is,
where
Results
Phylogenetic analysis of Zika strains
According to the organization of ZIKV genome, the complete coding region of ZIKV is first translated into a protein (polyprotein) which is then cleaved into three structural and seven nonstructural proteins. Based on the 60-dimensional natural vector method, the phylogenetic tree using 87 ZIKV polyproteins is reconstructed. As illustrated in Figure 1, the 87 ZIKV are well classified into three lineages: the Asian, West African, and East African lineages, which is consistent with the previous work (Lanciotti et al., 2016). The two African clades form a sister group to the Asian clade. The newly identified ZIKV strains in countries of Americas (such as Brazil, Haiti, Suriname, Guatemala, Martinique, and Puerto Rico) are all close to Asian and Pacific strains as shown in Figure 1.

Phylogenetic tree of 87 Zika viruses with polyproteins based on the 60-dimensional natural vector method. The strains are divided into three lineages: the Asian lineage, the West African, and East African lineages. Names, accession number, location, and year identified of the strains are displayed.
The strains from the Asian and Pacific countries are in the base of the Asian clade consisting of strains from Asia, Pacific regions, and Americas. This indicates that the epidemics in the Americas are likely dated back to strains from Malaysia isolated in 1966. The two African lineages are sister groups. The strains from Senegal are distributed in the two lineages, which suggest that two independent lineages have been circulating in this country. The MR766 prototype strains are clustered together as well.
Using 60-dimensional natural vectors, protein sequences coded by NS5 genes are employed to infer the phylogeny. NS5 is the largest protein in Zika and has the largest number of amino acid substitutions (Zhu et al., 2016). Thus, this protein provides more clues to evolutionary relationship between Zika strains. As displayed in Figure 2, the strains are grouped into two clades: the African 1 and African 2 clades. The recently emergent ZIKV strains in Americas are also near to those from Asia and Pacific islands. This result suggests that the Asian genotype expands to Americas. The tree also indicates that the Malaysia strains identified in 1966 are representative of an ancestral genotype for the Asian clade.
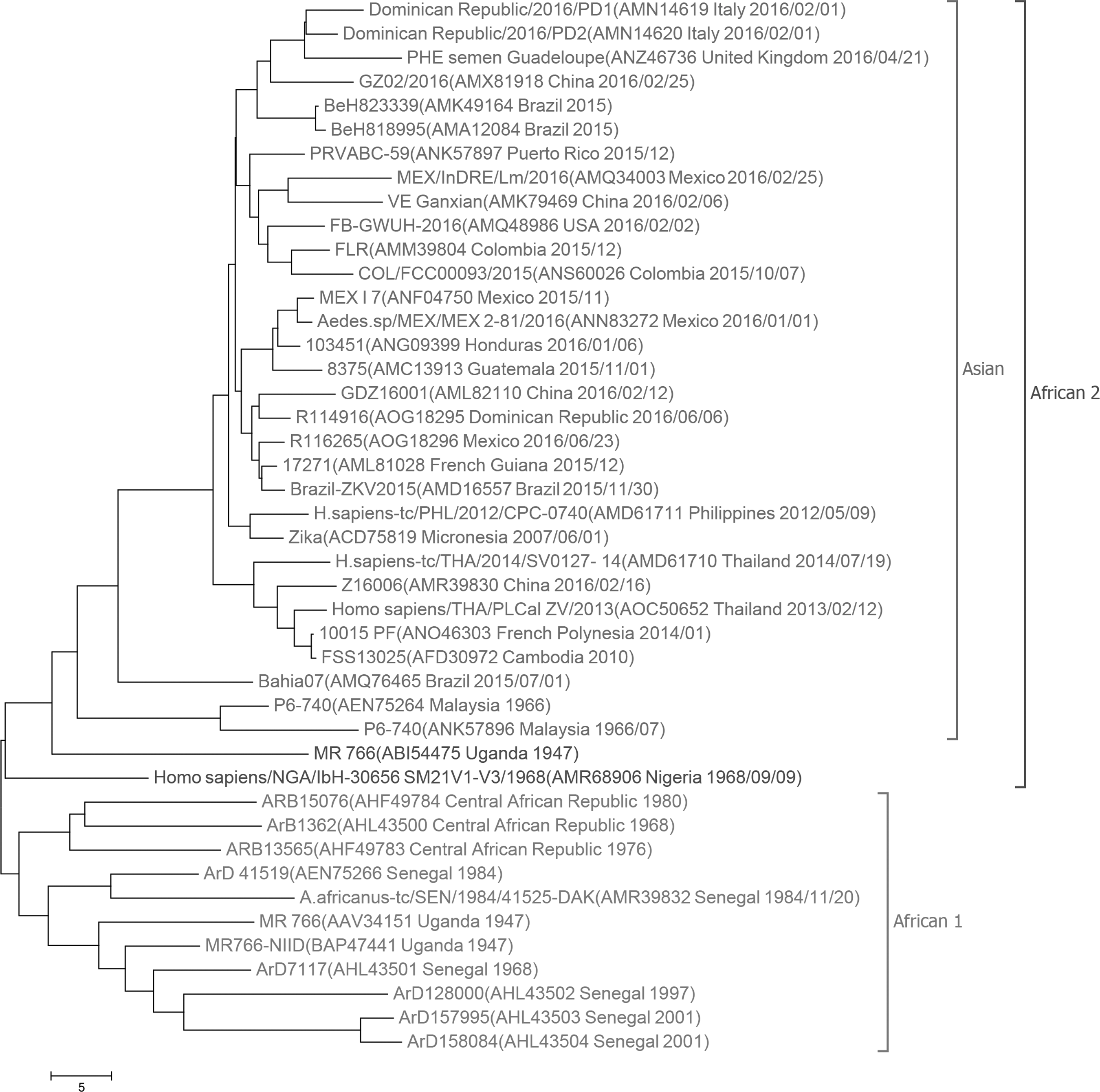
Phylogenetic tree of 44 Zika viruses with NS5 proteins based on the 60-dimensional natural vector method. The strains are divided into two lineages: the African 1 and African 2 lineages. Names, accession number, location, and year identified of the strains are displayed.
One MR766 strain and another strain from Nigeria are placed together with the Asian clade. The two African strains and those in the Asian clade are grouped together as a new clade, which is a sister group to other African ZIKV viruses. Thus, we conclude that the Asian clade is originated from Africa. The appearance of MR766 strains in the two African lineages shows their diversity at the beginning of isolation. Compared with the polyprotein phylogeny shown in Figure 1, the Zika strains in Africa are not divided into the West African and East African genotypes in Figure 2. In addition, MR 766 (ABI54475, Uganda 1947) and Homo sapiens/NGA/IbH-30656 SM21 V1–V3/1968 (AMR68906, Nigeria 1968/09/09) are near to the Asian group in the NS5 phylogenetic tree.
Applying the natural vector method, the phylogenetic tree of 87 complete coding regions of Zika strains is constructed. The tree is shown in Supplementary Figure S1. As illustrated in the figure, these strains are divided into the Asian group (in blue) and the African group (in red). The strain Homo sapiens/NGA/IbH-30656 SM21 V1-V3/1968 is closest to Aedes africanus/SEN/DakAr41524/1984 at distance 37.38. Compared with the polyprotein tree (Fig. 1), the West African strains are not separate from the East African strains. Using the NS5 gene, the phylogeny of 44 Zika strains based on natural vectors is shown in Supplementary Figure S2. It is obvious that the strains are clustered into the Asian clade (in green) and the African clade (in red). These two clades seem parallel. However, the NS5 protein phylogeny (Fig. 2) based on natural vectors indicates that the Asian strains originate from Africa. As a result, using the natural vector method, the polyprotein tree and the NS5 tree, both provide more insights into the origin and evolution of Zika strains than those based on DNA.
Phylogeny of flaviviruses
Using the natural vector method, the evolutionary history of 53 polyprotein sequences of flaviviruses is constructed as shown in Figure 3. The phylogenetic analysis suggests that the mosquito/vertebrate viruses form two separate lineages that are phylogenetically distant. ZIKV is placed together with Spondweni virus in one of the mosquito/vertebrate groups. The two dual host-affiliated insect-specific viruses, Donggang virus and Chaoyang virus, are positioned between the two lineages. The classical insect-specific flaviviruses are clustered forming one clade that includes Aedes flavivirus, Kamiti River virus, Cell fusing agent virus, Mercadeo virus, Parramatta River virus, Culex flavivirus, Quang Binh virus, and mosquito flavivirus.
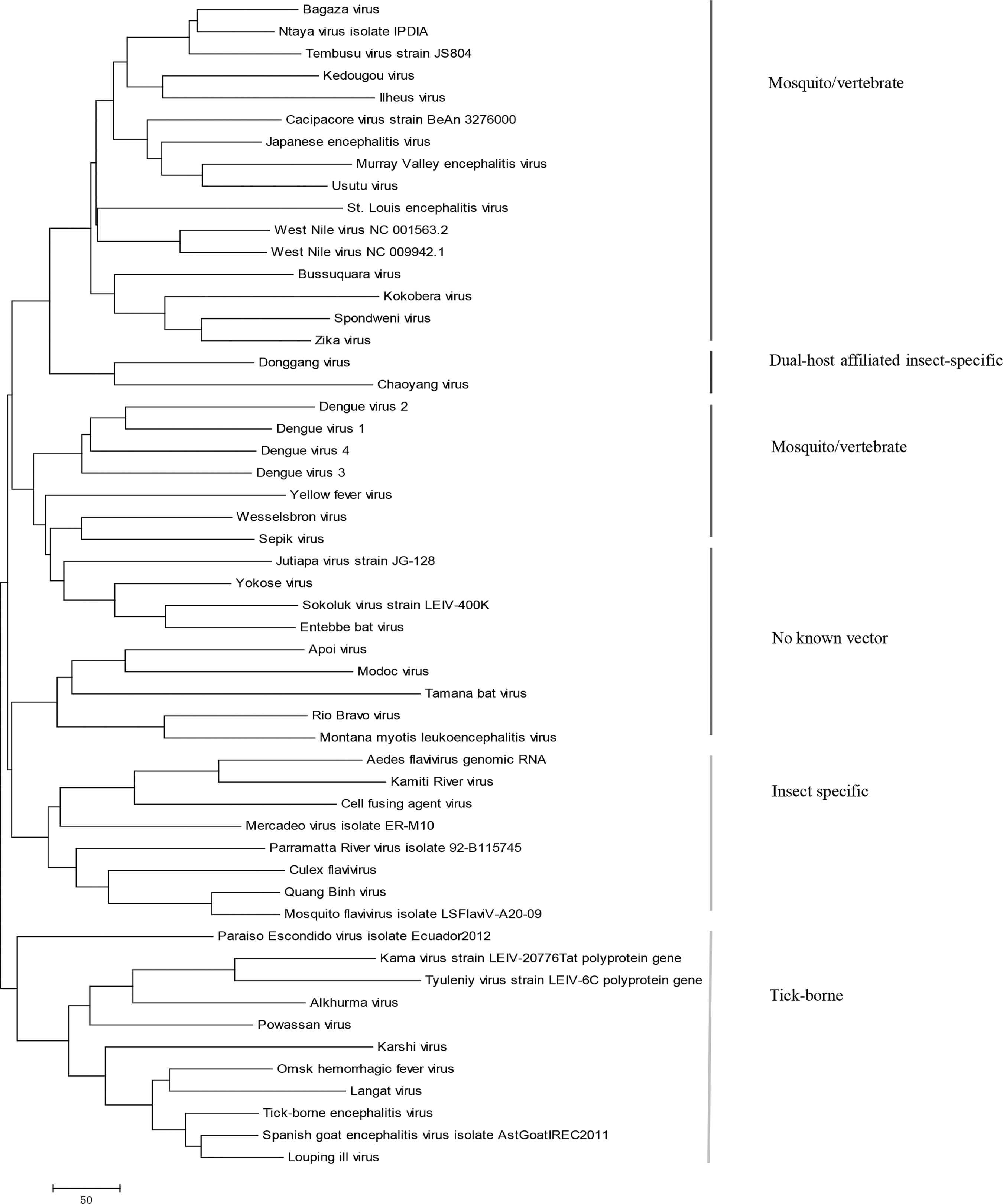
Phylogenetic tree of 53 viruses in the genus Flavivirus with polyproteins based on the 60-dimensional natural vector method.
The flaviviruses only infecting vertebrates with no known vector are separated into two clades. One clade is closely associated with the mosquito/vertebrate lineages. Jutiapa virus, Yokose virus, Sokoluk virus, and Entebbe bat virus belong to this clade. The other clade is distinctly different from other viruses and constitutes Apoi virus, Modoc virus, Tamana bat virus, Rio Bravo virus, and Montana Myotis leukoencephalitis virus. The viruses transmitted by ticks are grouped to form the tick-borne lineage consisting of Kama virus, Tyuleniy virus, Alkhurma virus, Powassan virus, Karshi virus, Omsk hemorrhagic fever virus, Langat virus, tick-borne encephalitis virus, Spanish goat encephalitis virus, and Louping ill virus. These results are consistent with the previous work (Huhtamo et al., 2014; Blitvich and Firth, 2015). The unclassified Paraiso Escondido virus is near to viruses in the tick-borne clade, and thus is predicted to belong to this group.
Discussion
In this work, we use the efficient natural vector method to infer the phylogeny of 87 ZIKV strains. This method is alignment-free and does not assume any parameters. Based on translated polyprotein sequences combined with the 60-dimensional natural vector, we classify the ZIKV viruses into three lineages: Asian, East African, and West African. The tree suggests that ZIKV evolves into three independent genotypes. It also indicates that the recent ZIKV infections in the Americas are imported from Asia and Pacific regions. Based on our phylogenetic analysis using NS5 protein in Figure 2, we first find that the ZIKV strains are evolving into two African lineages. This tree implies that the newly isolated strains in Americas originate from those in the Asian and Pacific islands. Our result demonstrates that ZIKV originates from Africa and expands into Asia, Pacific islands, and Americas. The MR766 strain (accession number: ABI54475) is one representative ancestral genotype for the Asian clade. Thus, we provide a novel evidence supporting the African origin hypothesis of ZIKV. This finding will help to track circulation of ZIKV in the world and elucidate the evolutionary history of ZIKV.
In Figure 1, the H. sapiens-tc/PHL/2012/CPC-0740 (AMD61711, Philippines) is nearest to the Asian genotype Aedes sp./MYS/P6-740/1966 (ANW07475) isolated in Malaysia in 1966. In Figure 2, however, the Philippines strain is closest to the Zika strain (ACD75819) isolated in Micronesia in 2007. The different position of the Philippines strain in two trees may be caused by the difference of mutation rate between the polyprotein and NS5 protein. The NS5 protein possibly has undergone higher mutation rate than the polyprotein. In addition, we also perform the phylogenetic analysis for the 53 viruses in the genus Flavivirus. The viruses are well classified into five groups. Each group is associated with typical transmission mode and host range. In particular, ZIKV is genetically near to Spondweni virus and both of them are positioned in one of the mosquito /vertebrate groups.
In the work of Lanciotti et al. (2016), three major lineages, Asian, West African, and East African lineages, are characterized. The result is based on the complete genomes and alignment. However, the number of ZIKV available used in their analysis is limited. The protein sequences of ZIKV are more conserved than DNA sequences and play important roles in viral activities. It may serve as markers to infer ZIKV evolution. Using complete protein sequences, the Zika strains are clustered into three clades, which is consistent with their result. In addition, all available ZIKV strains with full-length polyproteins are included in our analysis, which shows more diversity among the ZIKV strains.
In comparison, the phylogenetic tree based on alignment of NS5 proteins is built. The tree is obtained by ClustalW and the neighbor-joining algorithm implemented in MEGA 6.0. As shown in Figure 4, two major lineages, Asian and African lineages, are identified. However, the divergence time for the lineages is not clear. Using NS5 proteins, our phylogenetic tree construction by the natural vector method implies that ZIKV strains in Africa diverge earlier from the common ancestral than those in Asian clade. Compared with alignment-based methods using complete or partial genomes of ZIKV, our natural vector method using protein sequences provides novel insights into the origin, divergence, and evolution of ZIKV. Using the polyprotein sequences and multiple sequence alignment, a neighbor-joining phylogenetic tree of 53 flaviviruses is also plotted in Figure 5. Five clades are formed, which is in accordance with the tree derived from the natural vector method.
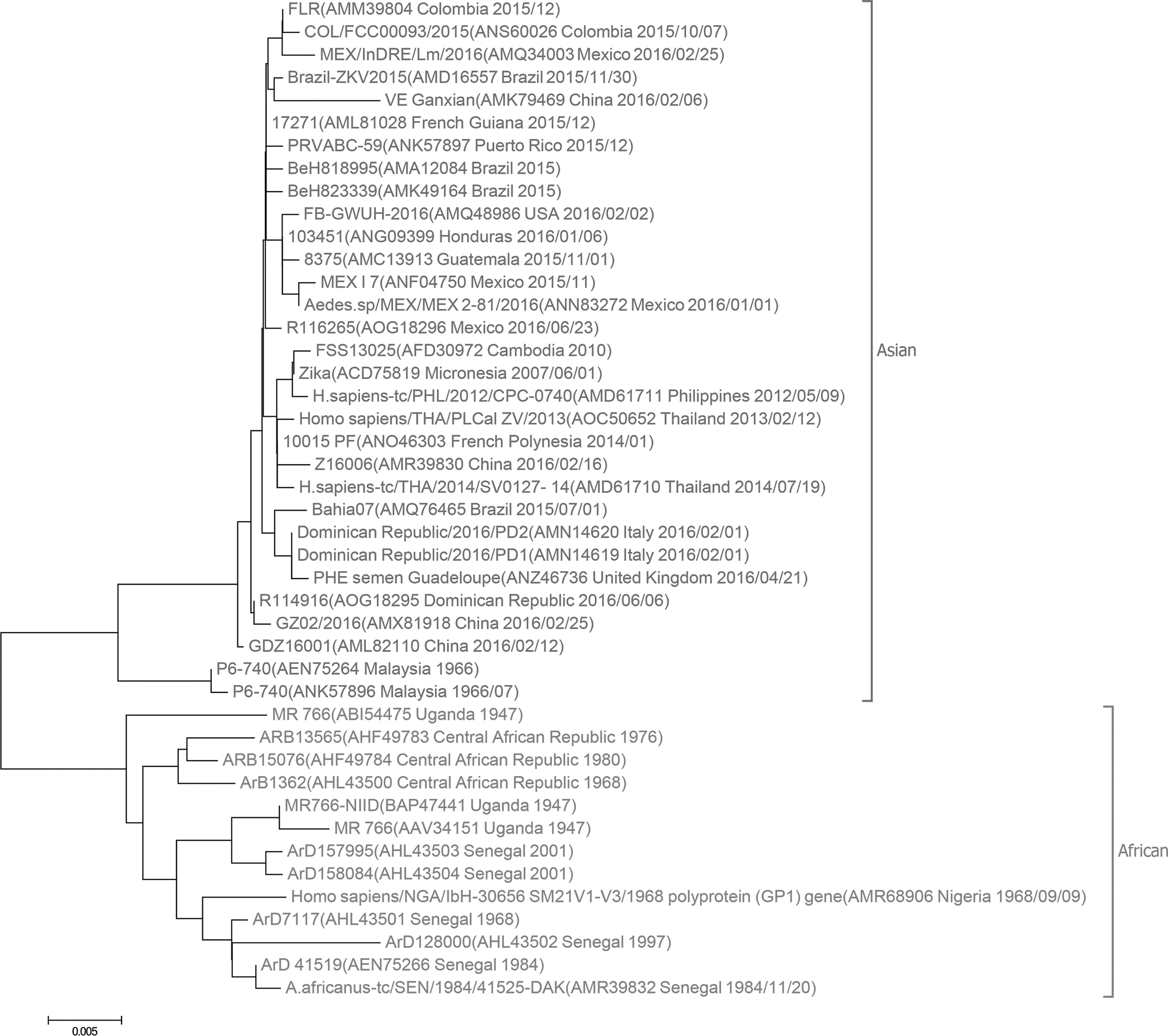
Phylogenetic tree of 44 Zika viruses with NS5 proteins based on multiple sequence alignment. The alignment is obtained by ClustalW and the tree is derived by neighbor-joining algorithm incorporated in MEGA 6.0.
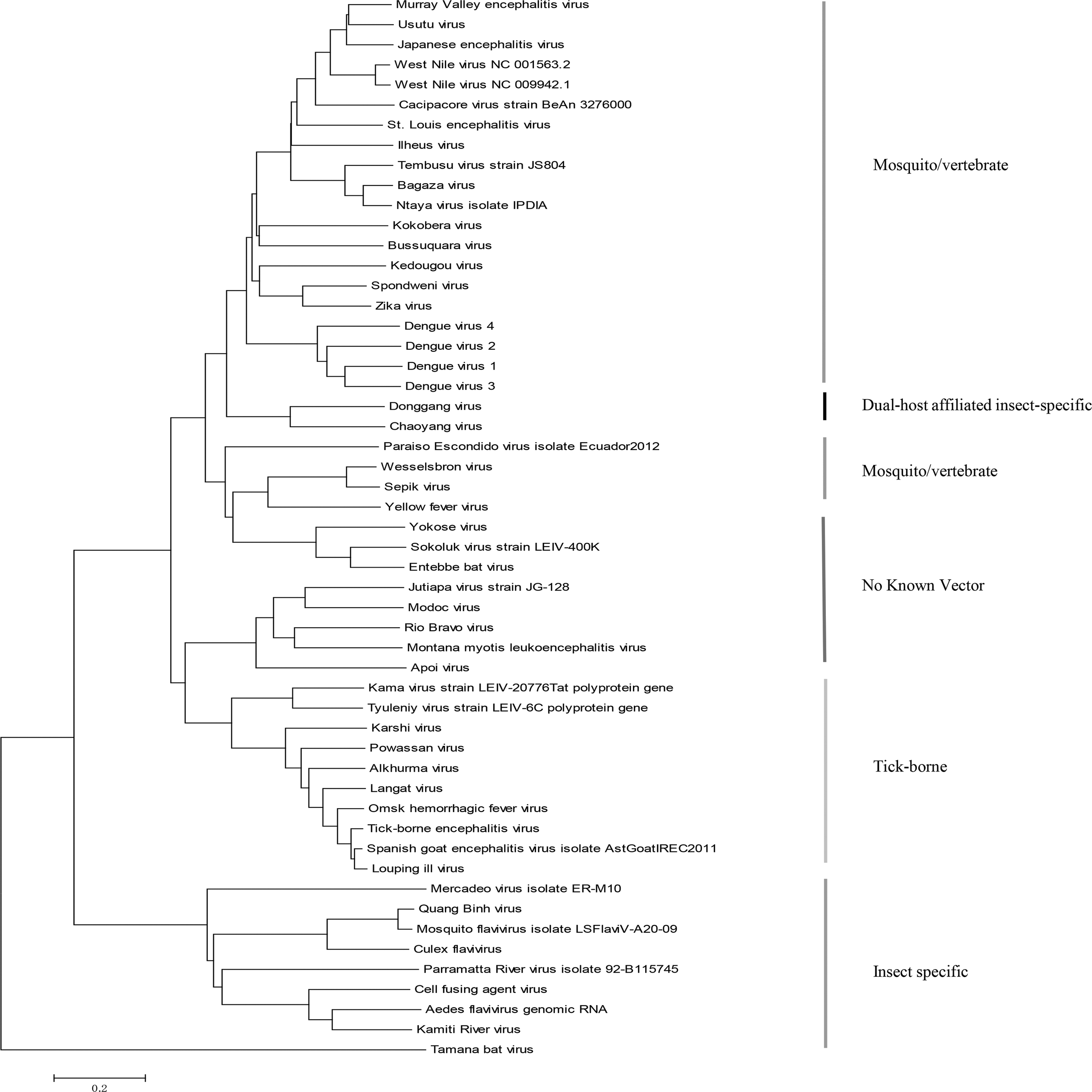
Phylogenetic tree of 53 viruses in the genus Flavivirus with polyproteins based on multiple sequence alignment. The alignment is obtained by ClustalW and the tree is derived by neighbor-joining algorithm incorporated in MEGA 6.0.
Various numerical representations of genetic sequences have been developed in the past decades. Recently, a two-dimensional (2D) graphical representation method is utilized to characterize Zika viruses (Nandy et al., 2016). Using the method, a genomic sequence of ZIKV is mapped to a 2D curve. For the curve, two parameters are defined to represent its center. The distance from the center to the origin denoting by g serves as sequence descriptor. However, the curve may have degeneracy and loops as well. The relationship between the sequence and the curve is not one-to-one. Moreover, the sequence descriptor may lose a lot of information of the curve. For example, MR 766 (HQ234498, Uganda 1947) has g = 84.13063 and its nearest strain is MEX I 7 (KX247632, Mexico 2015/11), where g = 84.13051. However, the two strains are from African and Asian genotypes, respectively. By our natural vector method, their distance is 69.404 and the nearest neighbor of MR 766 (HQ234498, Uganda 1947) is MR-766 (KX377335, Uganda 1947/04) with distance 5.459111.
Our numerical representation using 60-dimensional vectors is an efficient dimensionality reduction method for a long protein sequence. For each amino acid A, let nA
be the number of A,
One of the distinct advantages of natural vector is its efficiency. It is fast to convert sequences to numerical vectors and does not presume any model to infer phylogeny. The previous studies for ZIKV phylogeny are predominately generated by multiple sequence alignment that is very time consuming in computation. With the ClustalW algorithm implemented in MEGA 6.0 software, it takes 6 min to align the 44 NS5 protein sequences in a laptop with 8 GB RAM, Intel(R) Core(TM) i7-4500U CPU, and 1.80 GHz, while it only takes 0.26 s to convert these sequences to the natural vectors. Moreover, it needs 80 min to accomplish alignment for the 53 flaviviruses implemented by ClustalW, while the natural vector method only requires 1.09 s. Using our natural vector method, the classification of 776 single-segmented dsDNA viruses in the RefSeq database may be completed within 76.7 min on a PC computer (CPU 1.67 GHz, 3 GB of RAM). For multiple sequence alignment, this task is very difficult even infeasible in computation (Yu et al., 2013). According to the comparison using several examples, we conclude that the natural vector approach is a powerful tool to perform phylogeny analysis. It is accurate and competitive to alignment-based methods. It also has advantage over alignment-based methods in computational time.
Conclusions
In this study, we employ an alignment-free method, natural vector, to explore the evolutionary relationship among 87 ZIKV strains for the first time. The protein sequences of ZIKV are first introduced for phylogeny analysis. Based on the polyproteins, three major lineages are identified: the West African, the East African, and the Asian lineages. Applying the NS5 proteins, we find that the strains are divided into two African clades. This result confirms the hypothesis that ZIKV originates from Africa. Both the polyprotein tree and NS5 protein tree show that the ZIKV epidemic in Americas comes from Asia and Pacific islands. Our findings help to elucidate the origin and evolution of ZIKV. Besides, the phylogeny of 53 flaviviruses, including ZIKV, is constructed by the natural vector method based on polyproteins. In the tree, mosquito/vertebrate, dual host-affiliated insect-specific, classical insect-specific flaviviruses, tick-borne, and the no known vector groups are characterized. Especially, ZIKV is close to Spondweni virus belonging to the mosquito/vertebrate group. Our phylogenetic analysis of ZIKV and other flaviviruses may be helpful for the surveillance of these viruses and the vaccine design.
Footnotes
Acknowledgments
This study is supported by the National Natural Sciences Foundation of China (31271408 to S.S.-T.Y.), Tsinghua University startup fund (to S. S.-T.Y.), and Tsinghua University Independent Research Project Grant (to S. S.-T.Y.). The funders did not take part in study design, in collection and analysis of data, in the writing of the article, or in the decision to publish this article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.