
Abstract
The aim of this study was to investigate, for the first time, the effects of using adipose-derived mesenchymal stem cells (AD-MSCs) transfected with an episomal plasmid encoding fibroblast growth factor 1 (FGF1) (AD-MSCsFGF1), in providing the microenvironment required for angiogenic proliferation. The isolated rat AD-MSCs were positive for mesenchymal (CD29 and CD90) and negative for hematopoietic (CD34 and CD45) surface markers. Adipogenic and osteogenic differentiation of the AD-MSCs also occurred in the proper culture media. The presence of FGF1 in the conditioned medium from the AD-MSCsFGF1 was confirmed by Western blotting. G418 and PCR were used for selection of transfected cells and confirmation of the presence of FGF1 mRNA, respectively. Treatment with the AD-MSCFGF1-conditioned medium significantly increased the NIH-3T3 cell proliferation and human umbilical vein endothelial cell (HUVEC) tube formation compared to conditioned medium from nontransfected AD-MSCs (p < 0.001). In conclusion, the AD-MSCsFGF1 efficiently secreted functional FGF1, which promoted angiogenic proliferation. Using AD-MSCsFGF1 may provide a useful strategy in cell therapy, which can merge the beneficial effects of stem cells with the positive biological effects of FGF1 in various disorders, especially tissue defects, neurodegenerative, cardiovascular and diabetes endocrine pathologies, which remain to be tested in preclinical and clinical studies.
Introduction
F
In recent years, the gene-modified and gene-transfected mesenchymal stem cells (MSCs) have been proposed to enforce the paracrine effect of MSCs in producing the desired growth and trophic factors that were successful in some preclinical studies (Kurozumi et al., 2004). Most of these studies were conducted on BM-MSCs by viral vectors, which have their concerns about gene integration (Kim and Eberwine, 2010) such as fibroblast growth factor 2 (FGF2) (Ribeiro-Resende et al., 2012), hepatocyte growth factor (HGF) (Zhao et al., 2006), glial cell-derived neurotrophic factor (Horita et al., 2006), placental growth factor (Liu et al., 2006), and epidermal growth factor (EGF) (You and Nam, 2013). In recent years, a number of studies have determined FGFs and their receptors as key regulators in some vital biological functions, including senescence and self-renewal in a variety of cells (Coutu and Galipeau, 2011; Ribeiro-Resende et al., 2012). The first member of this family, FGF1 (acidic FGF), was described by Jaye et al. (1986). FGF1 is a multipotent growth factor and plays an important role in proliferation, differentiation, and cell survival (Rodriguez-Enfedaque et al., 2009). FGF1, however, binds to all known FGF receptors (Ornitz et al., 1996). Neurons predominantly express FGF1, which plays an important effect on neuroprotection and learning and memory functions. Rattus FGF1 as a single nonglycosylated polypeptide consisting of 154 amino acids with a molecular weight of 17 kDa (Zakrzewska et al., 2008) is also involved in angiogenesis, tissue repair, wound healing, adipogenesis, and homeostatic regulation (Walpurgis et al., 2011).
In this study, we aimed to use episomal plasmid vector FGF1 gene-transfected rat AD-MSCs for the first time, which can circumvent concerns about the viral vectors, including gene integration. We were also interested in determining whether FGF1-transfected AD-MSCs (AD-MSCsFGF1) would elaborate intact FGF1 that would accelerate fibroblast proliferation and human umbilical vein endothelial cells (HUVECs) tube formation.
Materials and Methods
Isolation and characterization of AD-MSCs
All surgical procedures were performed in accordance with the Animal Ethics Guidelines of Mashhad University of Medical Sciences (MUMS). Adult male Wistar rats (250–320 g) were obtained from the Animal Laboratory Facility, School of Medicine, Mashhad University of Medical Sciences, and were considered healthy donors for AD-MSCs.
Adipose tissue was collected using the following procedure: Rats were anesthetized using an intraperitoneal injection of ketamine hydrochloride (60 mg/kg) and xylazine hydrochloride (6 mg/kg), which were purchased from Rotex Medica (Trittau, Germany) and Alfasan (Woerden, Netherlands), respectively. Under aseptic conditions, a small surgical incision in the skin and muscular wall of left inguinal area was made and a small piece of subcutaneous fat pad was harvested and immediately placed into tissue culture tubes containing sterile phosphate buffer solution (PBS) with 100 IU/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO). Then, the animals were killed by an intracardiac injection of potassium chloride solution. Adipose tissue samples were minced carefully into very small pieces and washed with sterile PBS. Then, the tissue pieces were incubated in PBS containing 2 mg/mL of collagenase (Sigma-Aldrich) for 90 min at 37°C under constant shaking (Ghorbani et al., 2014a). The digested tissues were centrifuged at 2000 rpm for 5 min and the lipid layer was discarded. The pelleted stroma-vascular cells were washed twice with PBS and suspended in Dulbecco's modified Eagle's medium (DMEM; Gibco, Waltham, MA), which contained 10% fetal bovine serum (FBS; Gibco), 100 IU/mL penicillin, and 100 μg/mL streptomycin. The cells were seeded into the 25 cm2 flask and cultured under an atmosphere of 5% CO2 at 37°C. After 24 h, nonadherent cells were discarded by changing the medium and the adherent cells were expanded over three passages.
To confirm the presence of AD-MSCs, characteristic surface markers were evaluated using fluorescence-activated cell sorting (FACS) analysis (Secunda et al., 2015). The cells were incubated for 20 min at 4°C in the dark room with the following antibodies: CD90-phycoerythrin (PE) (NB100-65543PE; Novus Biologicals), CD29-fluorescein isothiocyanate (FITC, BD-561796; BD Biosciences, Franklin Lakes, NJ), CD45-FITC (bs-4819R; Bioss), and CD34b-PE (ABIN671373; Antibodies-Online). Matched isotype control antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). After washing with PBS, the cells were resuspended in 500 μL PBS containing 2% FBS and subjected to flow cytometric analysis using a FACS Calibur (BD Biosciences) flow cytometer. At least 1 × 104 cells per sample were acquired and analyzed.
Adipogenic differentiation of AD-MSCs
The AD-MSCs at passage 3 were seeded into 12-well plates at a density of 1 × 104 cells per well and cultured for 24 h in DMEM containing antibiotics and 10% FBS, and were then incubated with a differentiation medium consisting of DMEM supplemented with antibiotics, 3% FBS, 250 μM IBMX (Fluka, Switzerland), 1 μM dexamethasone, 5 μM indomethacin, 0.2 μM insulin, 34 μM D-pantothenate, and 66 μM biotin. The cells were maintained in this medium for 3 days and then cultured in an adipogenic maintenance medium consisting of DMEM supplemented with 3% FBS, 0.2 μM insulin, 1 μM dexamethasone, 34 μM D-pantothenate, and 66 μM biotin for 9 days (Yu et al., 2011; Feizpour et al., 2014). Adipogenesis was evaluated using the Oil-Red O staining method and photographed (Aguena et al., 2012).
Osteogenic differentiation of AD-MSCs
The AD-MSCs at passage 3 were seeded in 12-well tissue culture plates (1 × 104 cells/well) and cultured for 24 h in DMEM containing antibiotics and 10% FBS. Then, the medium was changed with an osteogenic differentiation medium consisting of DMEM supplemented with 10% FBS, 10 μg/mL ascorbic acid, 5 mM β-glycerol phosphate (Sigma-Aldrich), and 0.1 μM dexamethasone. The differentiation medium was replaced every 3 days and the cells were cultured in this medium for 2 weeks (Wang et al., 2009; Hsu et al., 2012). The osteogenic differentiation was evaluated by Alizarin red staining method. The cells were fixed with 10% formalin and exposed to 2% Alizarin red solution and photomicrographed (Ghorbani et al., 2014b; Pevsner-Fischer et al., 2012).
FGF1 transfection of AD-MSCs
AD-MSCs were seeded into the six-well plate with 2 mL DMEM (high glucose) containing 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% FBS and subjected to transfection when they reached 70% confluency at the passage 3. First of all, for evaluating the potential capability of transfection and determining the proper concentration of DNA and Lipofectamine, the AD-MSCs were transfected using different concentrations of pcDNA3.1-EGFP vector (0.5, 0.8, and 0.1 μg) using Lipofectamine® 2000 LTX and Plus™ Reagent (Gibco), according to the manufacturer's instruction. Then, using the same conditions, the AD-MSCs were transfected by 0.8 μg of the pCMV6-Entry vector with Myc-DDK-tagged ORF clone of Rattus norvegicus FGF1 (OriGene Technologies, Rockville, MD). The medium was changed 4 h after the start of transfection and then further replaced daily on consecutive days and stored at −80°C for FGF1 assessment. Nontransfected AD-MSCs were used as control cells.
Western blotting
The Western blot analysis for FGF1 was performed on the conditioned media collected from posttransfection day 6 because the preliminary analysis showed that its expression reached a maximum at this time point. Briefly, the conditioned media from AD-MSCs transiently transfected with the FGF1 (5 × 105 cells) were collected and the cell debris was removed by centrifugation. After that, the protein concentration was measured using the bicinchoninic acid protein assay kit (Sigma, St. Louis, MO) and equal amounts of protein from the samples (∼10 μg total protein) were mixed with the loading buffer. The samples were separated by electrophoresis in 12% sodium dodecyl sulfate –polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Santa Cruz Biotechnology). The membranes were then blocked with 5% skim milk and then blotted with the antibody specific to rat FGF1 (Abcam, Cambridge, MA) diluted in TBST buffer (10 mM Tris-HCl pH 7.5, 100 mM NaCl, and 0.1% Tween-20).
The bound antibody was made visible using horseradish peroxidase-conjugated goat anti-rabbit secondary antibody by enhanced chemiluminescence system (Bio-Rad, Hercules, CA) and detected using Gel Doc UV Alliance (UVItec Limited, Cambridge, United Kingdom). The resulting bands were quantified as OD × band area using NIH Image J software and expressed in arbitrary units. The conditioned media of nontransfected AD-MSCs were analyzed in parallel as control. Since there is no specific and well-accepted protein loading control for secreted protein, cell numbers and total protein were used for normalizing the conditioned medium for each transfectant subjected to FGF1 immunoblot analysis.
G418 sensitivity of AD-MSCs
G418 (Geneticin; Santa Cruz Biotechnology) was used for selection of the FGF1-transfected AD-MSCs (AD-MSCsFGF1). Third-passage AD-MSCs were incubated in 24-well plates (5 × 105 cells/well). At 80% confluence, G418 was added at final concentrations of zero (as control), 25, 50, 100, 300, 400, 500, 600, and 800 μg/mL to triplicate wells. The medium was changed every other day. The day (day 5) in which all the cells were dead was determined. The optimal concentration of G418 for selection (100 μg/mL) was defined as the lowest dose at which all the cells died (Moore et al., 2010; Ren et al., 2014).
Selection of transfected AD-MSCs (AD-MSCsFGF1)
Since it was found that the transfected cells significantly express FGF1 protein on day 6 posttransfection, it was assumed that Neo resistance gene of the vector is expressed in its peak level on day 6 posttransfection and selection of AD-MSCsFGF1 with G418 was started at this time point. Briefly, on sixth day posttransfection, the medium was replaced by DMEM containing G418 (100 μg/mL) and this treatment was continued for six consecutive days. The detached untransfected cells were removed by changing the medium every other day (Moore et al., 2010; Ren et al., 2014). The selected cells were then maintained and expanded in the G418-free DMEM and used for confirmation of FGF1 mRNA expression.
Confirmation of FGF1 mRNA expression in transfected AD-MSCs (AD-MSCsFGF1)
The presence of FGF1 coding mRNA in the selected AD-MSCsFGF1 was confirmed by PCR analysis using specific primers (Table 1) (Macrogen, Seoul, South Korea). Total RNA was extracted by the RNX-Plus kit (CinnaGen, Tehran, Iran) and used for cDNA synthesis (Fermentas, Waltham, MA) as described by the supplier. The cDNA was used as the template for FGF1 fragment amplification in PCR. The PCR product was resolved and analyzed on 2% agarose gel.
bp, base pair; FGF1, fibroblast growth factor 1.
Cell proliferation assay
The NIH-3T3 cells were seeded in 96-well plates (1 × 104 per well) containing DMEM with 10% FBS and grown for 24 h. After that, 100 μL conditioned medium from day 6 of AD-MSCsFGF1 were added to wells. Conditioned medium from nontransfected AD-MSCs in day 6 was used as a control. After a 24-h incubation, 20 μL of 3-(4,5-dimethylthiazol-2y)-2,5-diphenyl-2H-tetrazolium chloride (MTT; Sigma-Aldrich) were added to each well and cells were incubated for 4 h under 5% CO2 at 37°C. The supernatant was removed and 100 μL dimethyl sulfoxide were added to each well. Samples were shaken slowly to dissolve the resulting formazan dye and the absorbance was measured at 570 nm with a reference of 620 nm using a StatFAX303 plate reader.
Tube formation assay
The HUVECs, obtained from Pasteur Institute (Tehran, Iran), were cultured in Medium 199 supplemented with FBS (20%), 200 μg/mL endothelial cell growth supplement, 2 mM glutamine, penicillin (100 units/mL), and streptomycin (100 μg/mL). All cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Trypsin/EDTA (Gibco) solution was used to passage cells whenever they became confluent (Marin et al., 2001; Mortazavian et al., 2013). An aliquot of growth factor-depleted Matrigel (BD Bioscience) was thawed completely and then kept on ice. Then 50 μL of Matrigel were plated into 96-well plates at a horizontal level that permits the Matrigel to distribute evenly and incubated at 37°C for 30 min (Francescone et al., 2011). The cells (5 × 104) were seeded into each well and 100 μL of the conditioned medium from AD-MSCsFGF1 (at 6 days), were added to each well. The HUVEC cells cultured in the presence of the conditioned medium of nontransfected AD-MSCs at day 6 were considered the control. The plates were incubated for a further 24 h. The resulting endothelial tubes from five random fields in each well were photographed using a digital camera attached to an inverted microscope at 40 × magnification. The average of tubes length was quantified using Image J software and expressed as relative to the control. It was determined as the ratio of total capillary tube length in experimental cultures over the total capillary tube length in control.
Statistical analysis
The Statistical Package for the Social Sciences (SPSS) 16.0 (SPSS, Inc., Chicago, IL) was used for statistical analysis. Data were expressed as mean ± standard deviation. Student's t-test and the one-way analysis of variance were used for statistical analyses. A p-value less than 0.05 was considered statistically significant.
Results
Immunophenotypic analysis of MSCs
Surface antigens of AD-MSCs were analyzed by flow cytometry. The AD-MSCs were positive for MSC-associated markers CD29 and CD90, and consistently negative for hematopoietic markers CD34 and CD45 (Fig. 1).
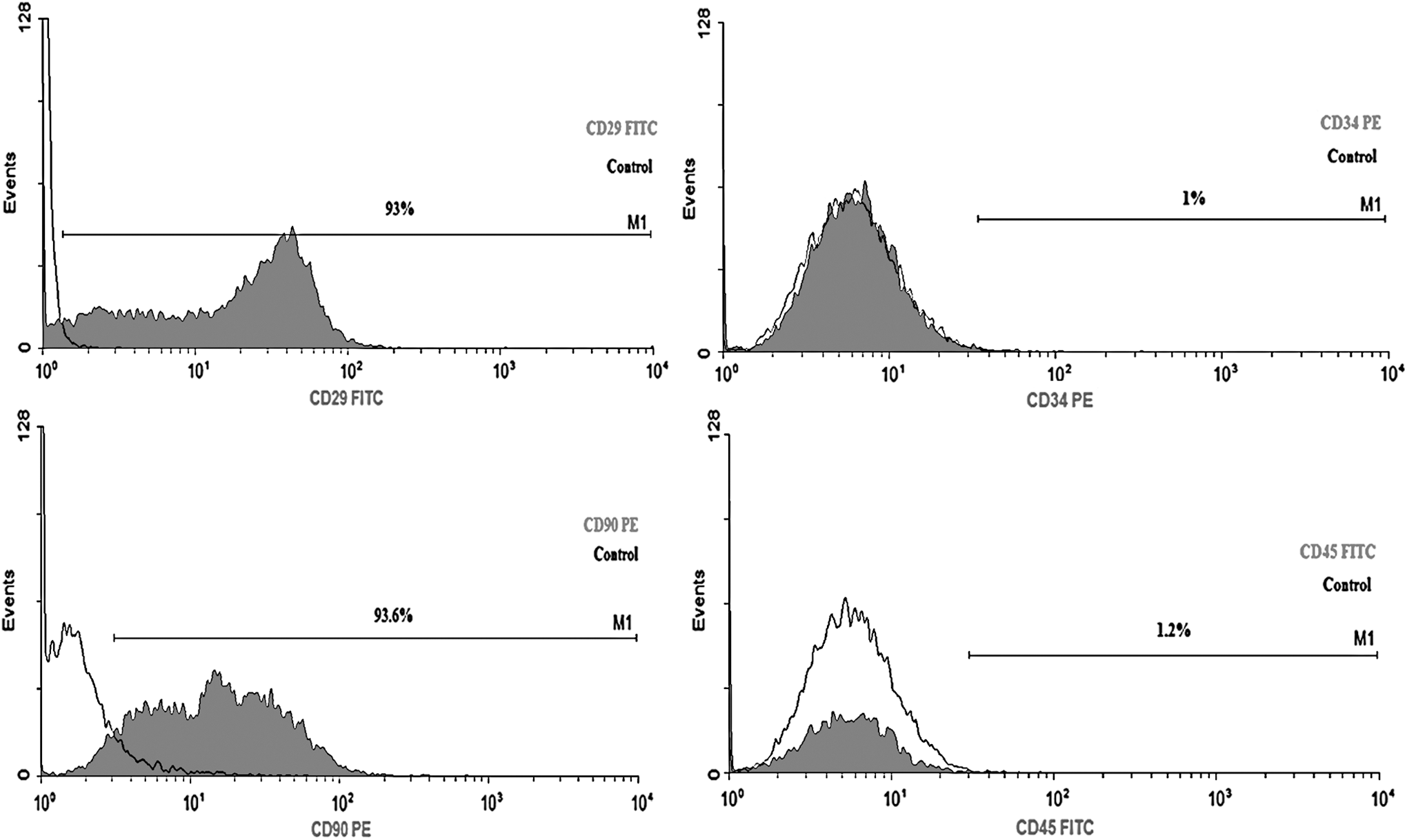
Flow cytometry characterization of AD-MSCs. MSCs expressed mesenchymal markers CD29 and CD90 (left) and were negative for hematopoietic markers, CD34 and CD45 (right). AD-MSCs, adipose-derived mesenchymal stem cells.
Adipogenic differentiation of MSCs
Figure 2 shows the adipogenic differentiation of AD-MSCs 9 days after treating with the adipogenic medium. The intracellular lipid droplets stained by Oil-Red O are visible.
Adipogenic differentiation of AD-MSCs. The AD-MSCs (third passage) were treated with adipogenic differentiation factors, as described in the Materials and Methods section. Then, the cells stained by Oil Red O staining (10 × ). Fat droplets are stained in the cytoplasm of the differentiated cells. The scale bar represents 100 μm.
Osteogenic differentiation of MSCs
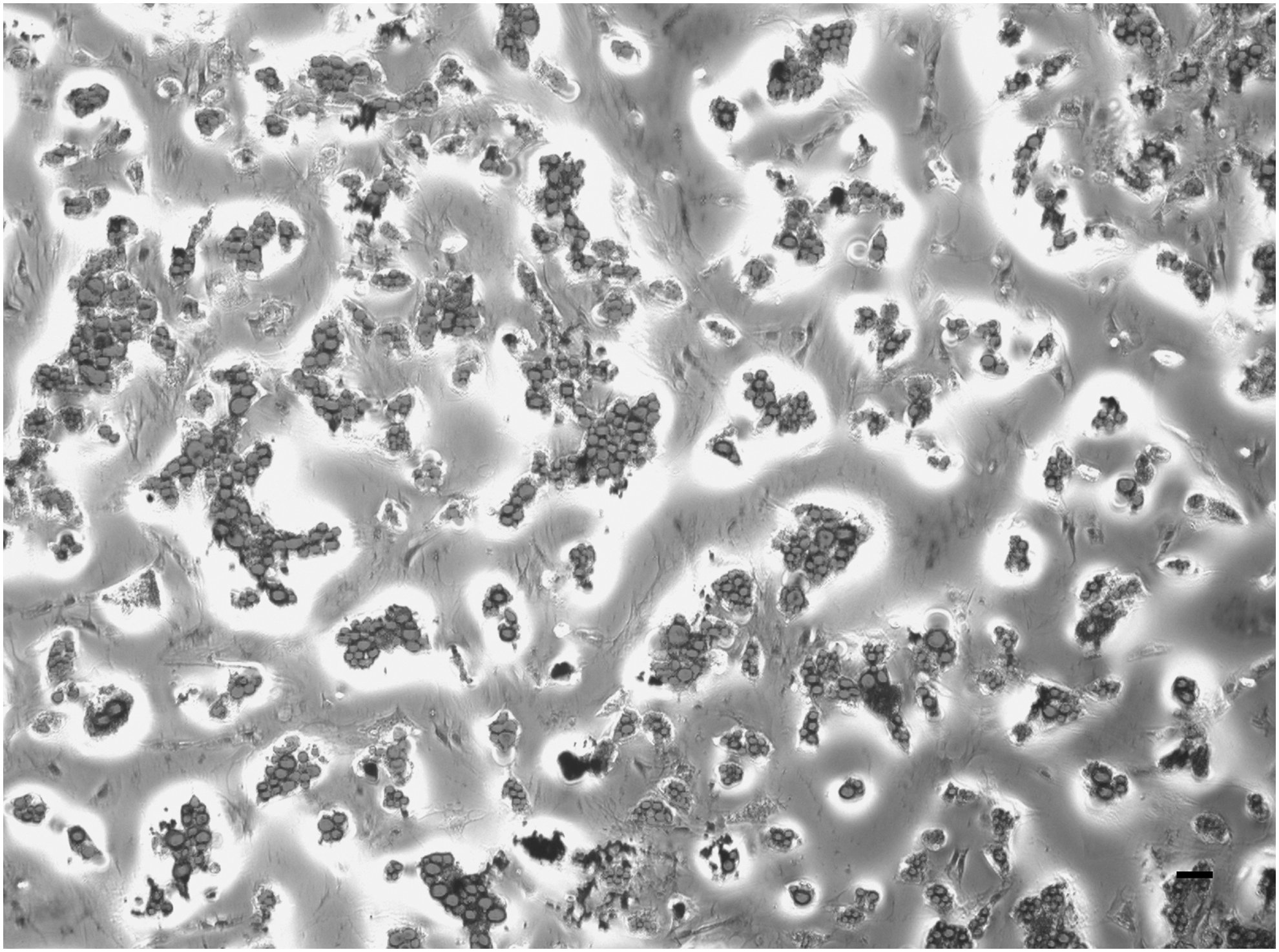
Figure 3 shows the osteogenic differentiation of AD-MSCs 2 weeks after treating with an osteogenic medium, which was evaluated by Alizarin red staining. The appearance of round polygonal cells with bright orange-red calcium deposits stained by Alizarin red confirmed the successful differentiation of AD-MSCs into osteoblastic cells.

Osteogenic differentiation of AD-MSCs. Differentiation was stimulated by treating AD-MSCs (third passage) with the osteogenic differentiation medium for 2 weeks and stained using Alizarin red. Calcium deposits can be seen as dark areas (arrows) (40 × ). The scale bar represents 100 μm.
Transfection of AD-MSCs by pcDNA3.1-EGFP vector
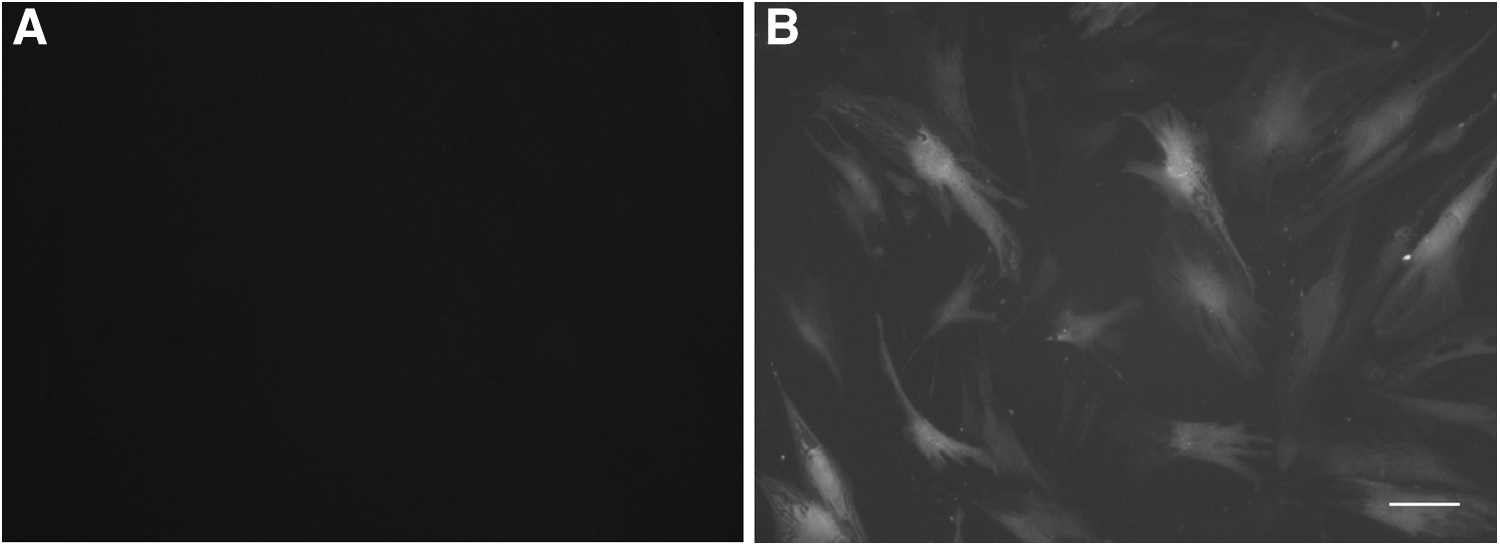
Figure 4B shows the presence of GFP (green fluorescent protein) in pcDNA3.1-EGFP transfected AD-MSCs. The best result for EGFP transfection and expression was obtained by using 0.8 μg pcDNA3.1-EGFP and 2 μL of lipofectamine 2000 per each well. These proportions were used to transfect AD-MSCs with a pCMV6-Entry vector containing FGF1 gene. Figure 4A shows the control group (nontransfected AD-MSCs).
FGF1 expression significantly increased on sixth day posttransfected AD-MSCs with FGF1 gene
From the day 6 posttransfection, a protein band of ∼17 kDa corresponding to rat FGF1 was confirmed by Western blotting (Fig. 5A). As shown in Figure 5B, there was a significant increase of FGF1 protein in conditioned media from the AD-MSCsFGF1 at day 6 posttransfection, compared with control AD-MSCs.

FGF1 protein expression
Selection of transfected AD-MSCs (AD-MSCsFGF1)
All the nontransfected AD-MSCs were completely detached from the plate when cultured in the presence of 100 μg/mL G418 within 5 days (Fig. 6). This dose was used for selection of AD-MSCsFGF1. The cells were then harvested on day 12 and used for assessment of FGF1 mRNA expression.

Determination of minimum lethal dose of G418 on AD-MSCs. The AD-MSCs were completely detached from the plate when cultured in the presence of 100 μg/mL G418 within 5 days. The lowest dose (100 μg/mL) was determined as optimal concentration for selection. The scale bar represents 200 μm.
The presence of FGF1 mRNA in the selected AD-MSCsFGF1 was confirmed by PCR
As shown in Figure 7, there was a 102 bp product that was amplified for the cDNA prepared from the AD-MSCsFGF1. It corresponds to the size of the amplicon expected to be amplified using the specific primers for Rattus FGF1. The cDNA from nontransfected AD-MSCs was used as negative control.
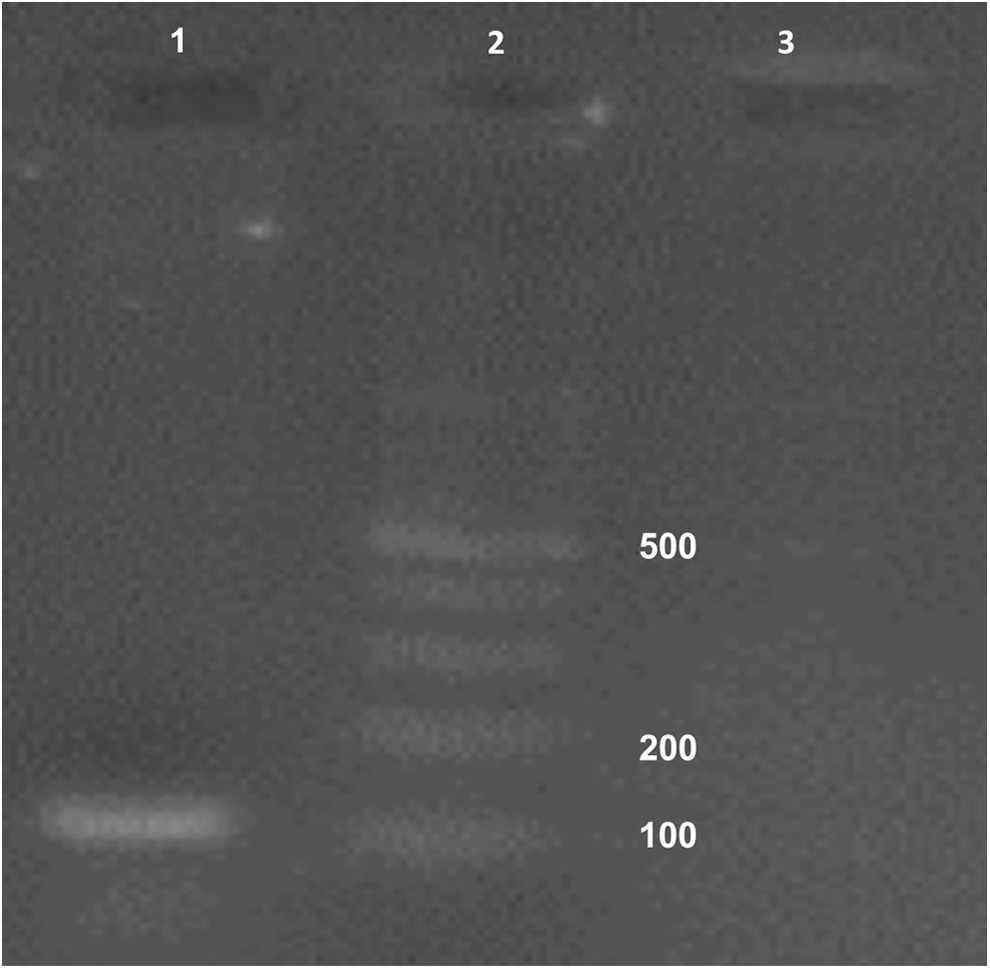
PCR confirmed the presence of Rattus FGF1 mRNA in transfected AD-MSCs. Lane 1: Expected band (102 bp) in transfected AD-MSCs; lane 2: Gene ruler (100 bp; Fermentas, Waltham, MA); lane 3: Nontransfected AD-MSCs as negative control.
Conditioned media from AD-MSCsFGF1 significantly increased proliferation of fibroblast cells
The NIH-3T3 cells were incubated for 24 h with the conditioned medium obtained from AD-MSCsFGF1 at sixth day posttransfection. Figure 8A and B show the microscopic feature of cells treated with AD-MSC- or AD-MSCFGF1-conditioned medium. As shown in Figure 8C, the NIH-3T3 cell proliferation treated with sixth day posttransfection AD-MSCsFGF1 was significantly increased (about 70%) compared to cells treated with nontransfected AD-MSC-conditioned medium (p < 0.001).
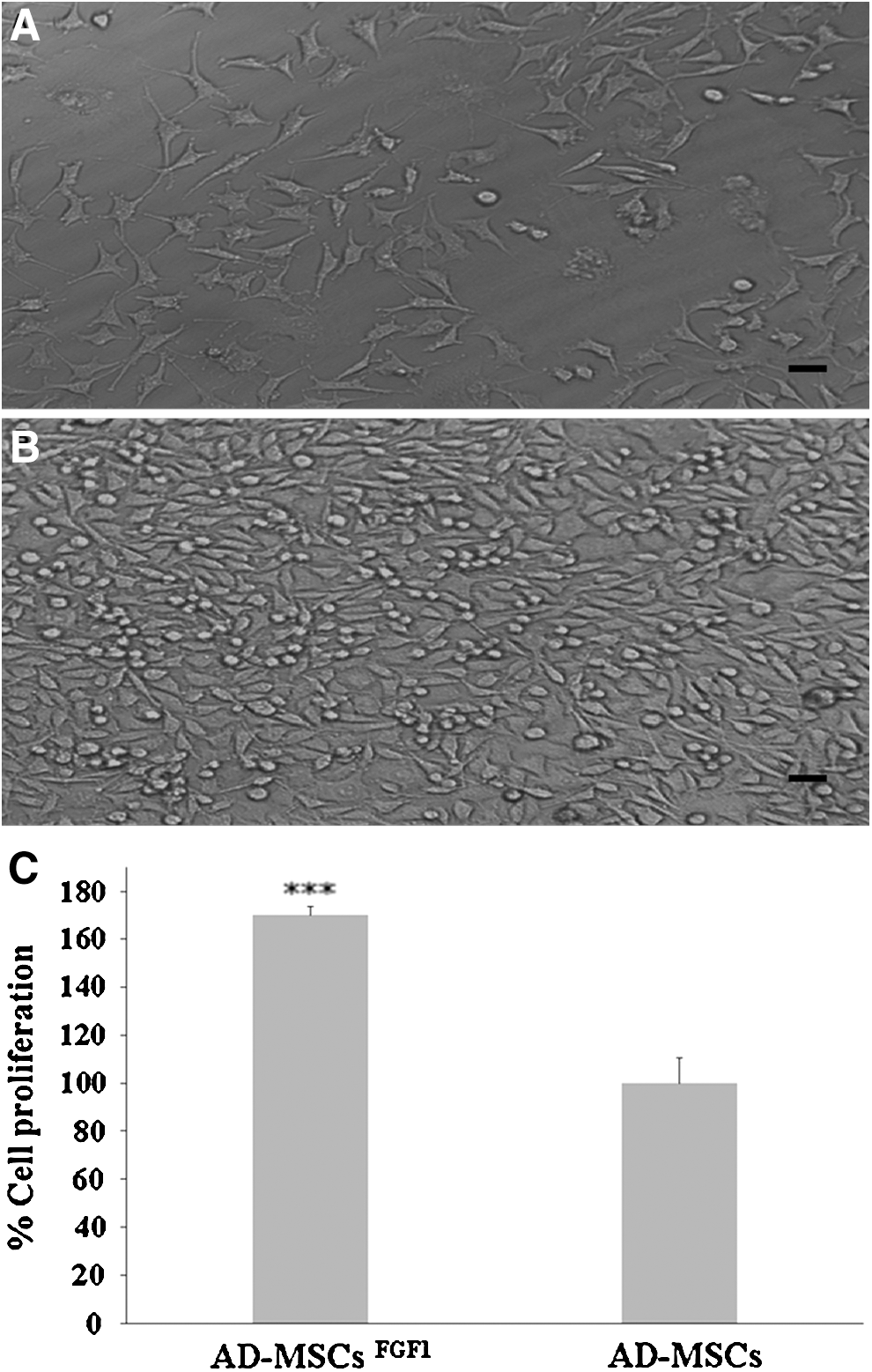
NIH-3T3 cells proliferation after 24 h incubation with condition media of AD-MSCs
Conditioned media from AD-MSCsFGF1 significantly increased tube formation by HUVECs
Figure 9A and B show the HUVECs 24 h after being treated with day 6 conditioned media of AD-MSCs and AD-MSCsFGF1, respectively. According to Figure 9C, there was an ∼4-fold increase in tube formation by HUVECs treated with day 6 conditioned media of AD-MSCsFGF1 in comparison to those treated with AD-MSCs (p < 0.001).
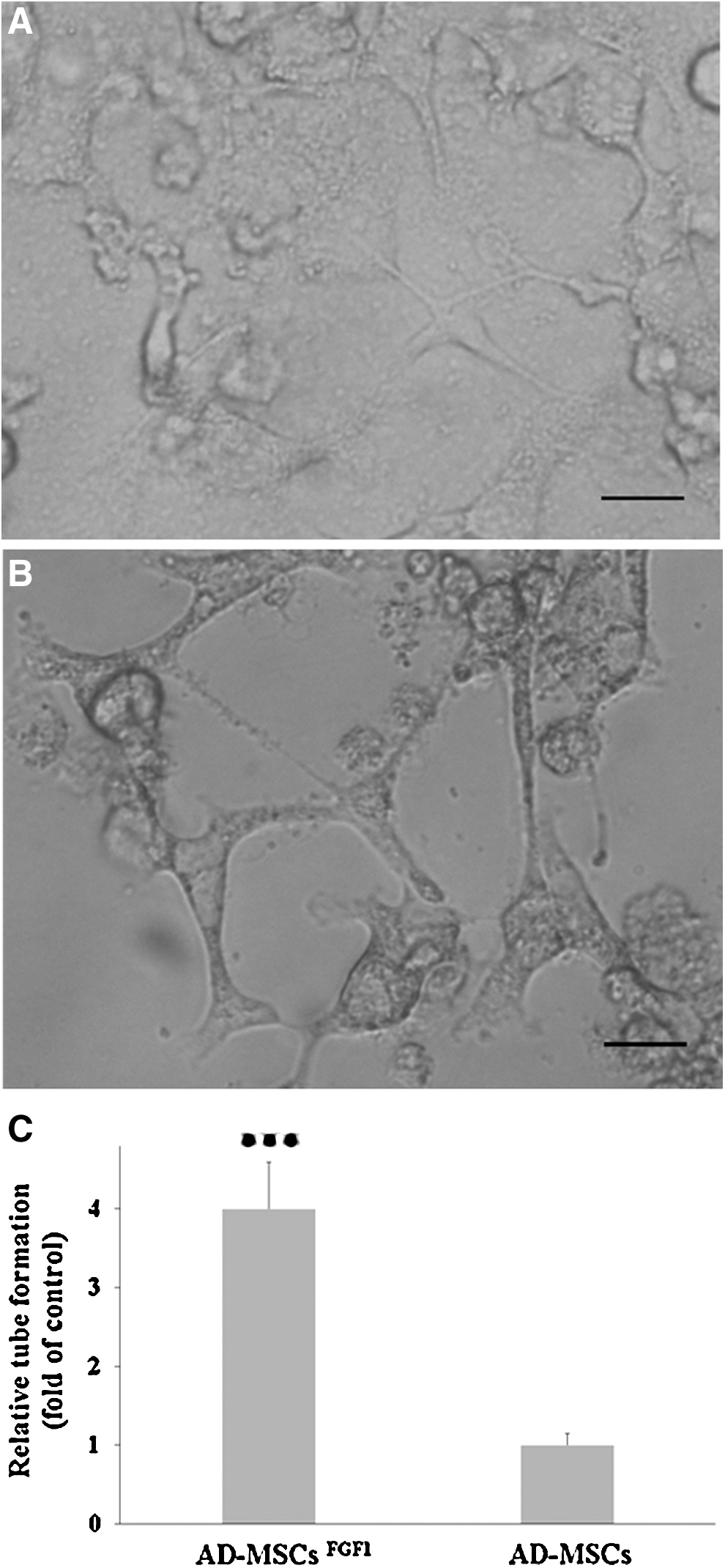
Tube formation by HUVECs treated 24 h with condition media of AD-MSCs
Discussion
In this study, we isolated and characterized rat AD-MSCs and successfully transfected them using a nonviral pCMV6-Entry vector containing rat FGF1 gene, as a transfection-ready DNA. The results of this study showed that after AD-MSC transfection, FGF1 mRNA and protein expression levels were significantly upregulated to levels that can be detected. The presence of FGF1 in AD-MSCFGF1-conditioned medium was confirmed by Western blotting on day 6 posttransfection. The functionality of secreted FGF1 was also confirmed by increased NIH-3T3 cell proliferation and HUVEC tube formation.
Although FGF1 lacks a signal sequence and is normally located in the cytoplasm or the nucleus, it is secreted by an alternative exocytotic mechanism that bypasses the endoplasmic reticulum–Golgi pathway (Alzheimer and Werner, 2002), which is consistent with the presence of FGF1 in the AD-MSCFGF1-conditioned medium, in our study. Since human and rat FGF1 are very similar in structure (96.4% sequence identity), the tube formation assay was conducted on HUVECs (Kulahin et al., 2007). In contrast to the AD-MSC-treated group, the HUVECs treated with AD-MSCFGF1-conditioned medium were very well organized into tube-like structures. FGF1 is a multipotent factor and can promote proliferation, differentiation, and survival in a wide variety of cells of mesodermal and neuroectodermal origins (Bryckaert et al., 2000; Rodriguez-Enfedaque et al., 2009). Our results demonstrated a significant increase in proliferation of fibroblast NIH-3T3 cells treated with AD-MSCsFGF1-conditioned medium, which is in accordance with other studies (Bryckaert et al., 2000; Siffroi-Fernandez et al., 2005). FGF1 can also play a critical role in angiogenesis (Mori et al., 2013). It is a more potent angiogenic agent in comparison to vascular endothelial growth factor (VEGF) or platelet-derived growth factor (Yun et al., 2010). FGF1 can also induce the physical organization of endothelial cells into tube-like structures (Javerzat et al., 2002), which is consistent with our tube formation data.
Therapeutic tissue regeneration depends on two critical requisites: a reliable source of stem cells and cytokine growth factors as signals. Compared to other types of stem cells, adipose-derived stem cells exhibit several advantages. These cells can be easily harvested from autologous fat tissue with high yield and by minimally invasive procedures, and their use was not limited by immunogenicity, ethical and political issues (Baer and Geiger, 2012; Jun-Jiang and Huan-Jiu, 2016). Self-renewal for long periods and differentiation into mesenchymal lineage cells such as osteoblasts, adipocytes, chondrocytes, and myocytes (Rodriguez et al., 2006; Lin et al., 2008), as well as transdifferentiation into neurons, neurotrophic factor secreting cells, Schwann cells, and endothelial and epithelial cells (Lin et al., 2008; Shi et al., 2012; Derby et al., 2014) make these cells suitable for regenerative medicine relying on cell and gene therapies (Bunnell et al., 2008; De Francesco et al., 2015).
AD-MSCs also express several kinds of growth factor receptors and by secretion of various cytokines and growth factors such as interleukin (IL)-6, IL-8, IL-10, HGF, VEGF, or FGF2, exhibit immunomodulatory, angiogenic, and antiapoptotic properties, in vitro or in vivo (Kilroy et al., 2007; De Francesco et al., 2015). AD-MSCs can act to suppress the proliferation and function of activated lymphocytes, inhibit the production of proinflammatory cytokines, and induce regulatory T cells (Patel et al., 2008; Gonzalez-Rey et al., 2010; Nagaya et al., 2014). AD-MSCs are also capable to reduce the damage, promote regeneration, or restore function of different tissues such as nervous system, heart, liver, or pancreas (Salgado et al., 2010; Zack-Williams et al., 2015; Bajek et al., 2016). In addition, it has been shown that AD-MSCs are prone to be introduced through exogenous genes and used as vehicles for delivering or producing beneficial proteins for therapeutic purposes (Song et al., 2012; Jun-Jiang and Huan-Jiu, 2016). Therefore, AD-MSCs can be regarded as a promising approach for stem cell-based therapy.
While the physiological role of FGF1 in not established and FGF1 knockout mice are apparently completely normal, viable, and fertile (Miller et al., 2000), the therapeutic potentials of FGF1 in angiogenesis (Uriel et al., 2006), cytoprotection, and tissue repair such as wound healing and bone formation (Steiling and Werner, 2003; Kelpke et al., 2004; Blaber et al., 2015), adipose remodeling, and metabolic homeostasis (Jonker et al., 2012) have been suggested. Several investigations indicated that FGF1 could induce myocardial angiogenesis and arteriogenesis, and improve cardiac function by recruitment of progenitor cells, increased mitosis, and decreased apoptosis of cardiomyocytes in ischemic heart disease (Cuevas et al., 1997; Engel et al., 2006; Formiga et al., 2014). In the same way, the overexpression of myocardial FGF1 has been shown to induce differentiation and growth of the coronary artery system (Fernandez et al., 2000) and enhance cardiac functional recovery and cell survival during ischemia and reperfusion injury (Palmen et al., 2004). In addition, its expression was increased in the pericardial fluid of patients with cardiac ischemia (Iwakura et al., 2000). Myocardial neoangiogenesis was also increased following intramyocardial injection of FGF1 in patients with coronary artery disease (Schumacher et al., 1998). Moreover, FGF1, probably by mechanisms other than angiogenic induction, improved perfusion and reduced the risk of amputations in patients with critical limb ischemia (Comerota et al., 2002; Nikol et al., 2008).
FGF1 is also extensively expressed in nervous tissue. In the CNS, FGF1 is localized predominantly in sensory and motor neurons and also released from astroglial cells in response to stress (Ford-Perriss et al., 2001; Di Serio et al., 2005; Matsunaga and Ueda, 2006; Meyer, 2014). The role of FGF1 in neurogenesis, neurite outgrowth, and neuronal maturation and differentiation is well described (Turner et al., 2006; Hsu et al., 2013). FGF1 has been shown to act as a neurotrophic, neuroprotective, and angiogenic factor during development and repair process following brain or spinal cord injury (Hossain et al., 2002; Dono, 2003; Cheng et al., 2011; Huang et al., 2011; Tsai et al., 2015). Local administration of FGF1 caused significant functional improvements in obstetric brachial plexus palsy and chronic transverse myelitis (Lin et al., 2005, 2006). FGF1 abnormality has also been shown to be associated with neurodegenerative (such as Alzheimer's disease) and demyelinating diseases (Mashayekhi et al., 2010; Zhou et al., 2012; Tao et al., 2014). An interesting recent study also showed that recombinant FGF1 induces potent, insulin-dependent glucose lowering in diabetic mice that is dose dependent, but does not lead to hypoglycemia (Suh et al., 2014). However, FGF1 has poor stability and relatively short half-life because is readily inactivated or degraded by diffusional loss and/or proteases following directly in vivo administration (Zakrzewska et al., 2008).
Although many types of delivery systems have been proposed to carry FGF1 and improve their therapeutic efficacy, in vitro or in vivo, more efficient delivering systems still remain to be developed (Yun et al., 2010). Our described AD-MSCsFGF1 could potentially be used as an FGF1 delivery system to merge the useful biological functions of FGF1 with the beneficial therapeutic effects of AD-MSCs. It could be considered a promising approach for future therapeutic application against various disorders especially tissue defect and neurodegenerative, cardiovascular, and diabetes endocrine pathologies, which remain to be tested in preclinical and clinical studies.
There were a number of limitations in this study. First, the long-term safety of AD-MSCs encoding FGF1 gene was not determined. Increased mitogenesis, angiogenesis, and cell survival by FGF1 may contribute either to the onset or to faster progression of cancer (Zakrzewska et al., 2008). Second, we have not considered the adipogenic differentiation of AD-MSCsFGF1. FGF1 has been suggested as a key factor in adipose remodeling. Hutley et al. showed that adipose-derived microvascular endothelial cells secrete FGF1, which has a proadipogenic activity on human preadipocytes, in vitro (Hutley et al., 2004). Our study was involved with the isolation of AD-MSCs cells from the stromal vascular fraction (SVF) of homogenized adipose tissue. SVF represents a heterogeneous population of cells, including CD34+/CD31− cell fraction, that is, the preadipocytes (Sengenes et al., 2005). However, the adherence to plastic and subsequent successive passages results in a relatively homogenous cell population expressing stem cell markers, compared with the heterogeneity of the SVF (Mitchell et al., 2006). Moreover, the preadipocytes comprise as little as 0.02% of the SVF and have a limited life-span (Zuk et al., 2001). On the other hand, it was shown that the AD-MSCs can enter the adipose lineage at a high rate and differentiate into cells that resemble the properties similar to adipocytes (Villageois et al., 2012). From the other point of view, mice with an FGF1 deficiency displayed a severe diabetic phenotype coupled to aberrant adipose expansion in response to high-fat diet (Jonker et al., 2012). It was shown that pharmacological administration of recombinant FGF1 effectively decreased glucose level, insulin resistance, and hepatic steatosis (by stimulating hepatic lipid catabolism) in genetically and diet-induced obese mice (Suh et al., 2014; Liu et al., 2016).
Nevertheless, this study still introduces a system for cellular therapy, in which the transfected stem cells efficiently secrete functional FGF1, which may increase the functional half-life and also the local concentration of the FGF at the site of homing.
Conclusions
FGF1 regulates many biological functions such as survival, cell proliferation, migration, and differentiation. FGF1 gene-transfected AD-MSCs can propose a suitable strategy, which may provide an opportunity to combine these unique properties of FGF1 with the therapeutic potential of AD-MSCs for cell therapy in many conditions, including ischemia-caused cerebrovascular and cardiovascular disorders.
Footnotes
Acknowledgments
The authors would like to thank the Vice Chancellery for Research and Technology and the Stem Cell Research and Application Core of Mashhad University of Medical Sciences, and the Iran's Vice-Presidency for Science and Technology (Grant No. 11/63941) for financial supports. The results described in this article were extracted from Seyed Javad Hoseini's PhD thesis (Grant No. 910807) and Hamed Ghazavi's PhD thesis (Grant No. 921716) at Mashhad University of Medical Sciences, Mashhad, Iran.
Disclosure Statement
No competing financial interests exist.