
Abstract
Leukotriene B4 receptor 1 (BLT1), a high-affinity G protein-coupled receptor (GPCR) for leukotriene B4 (LTB4), plays important roles in inflammatory and immune reactions. Although the LTB4–BLT1 axis is known to promote inflammation, the binding proteins that modulate LTB4–BLT1 signaling have not been identified. Recently, we discovered that receptor for advanced glycation end products (RAGE) interacts with BLT1 and modulates LTB4–BLT1 signaling. We propose RAGE as a new class of GPCR modulator and a new target of future GPCR studies.
Introduction
L
Receptor for advanced glycation end products (RAGE) is a member of the immunoglobulin superfamily (Neeper et al., 1992) and is involved in several inflammatory diseases, such as diabetes, atherosclerosis, and neurological disorders (Forbes et al., 2004; Vincent et al., 2007; Soro-Paavonen et al., 2008). Recent studies demonstrated that RAGE interacts with other receptors, including toll-like receptors (TLRs) and several GPCRs through direct interaction with downstream adapter molecules or receptors. TIRAP and MyD88, which are adaptor proteins for TLR2/4, bind to phosphorylated RAGE and transduce a signal to downstream molecules, including IRAK4 (Sakaguchi et al., 2011). RAGE physically interacts with formyl peptide receptor 1 (FPR1) and formyl peptide receptor-like 1 (FPRL1), which are GPCRs for formyl peptides and share significant homology with BLT1, and influences downstream signaling, including extracellular signal-regulated kinase (ERK) 1/2 phosphorylation and cyclic adenosine monophosphate (AMP) accumulation (Slowik et al., 2012).
Recently, we revealed that RAGE acts as a novel binding protein for BLT1 (Ichiki et al., 2016). We also showed that RAGE modulates LTB4–BLT1 signaling and affects cell migration through BLT1 both in vitro and in vivo. In this report, we discuss in detail the role of RAGE in BLT1 signaling.
RAGE Enhances LTB4–BLT1-Dependent ERK Phosphorylation and Subsequently Suppresses NF-κB Activation
We demonstrated for the first time that RAGE interacts with BLT1 and enhances LTB4–BLT1-dependent ERK phosphorylation. RAGE inhibited LTB4–BLT1-dependent NF-κB activation and upregulation of proinflammatory cytokines and chemokines. This RAGE-dependent inhibition of NF-κB was blunted by inhibition of MEK, a proximal upstream kinase of ERK, suggesting that RAGE suppresses LTB4–BLT1-dependent NF-κB signaling by enhancing signaling through the MEK–ERK pathway.
Although the relationship between NF-κB activation and the MEK–ERK pathway seems controversial, several reports have also demonstrated that the MEK–ERK pathway negatively regulates NF-κB activation by affecting phosphorylation of the p65 NF-κB subunit or TATA-binding protein, an NF-κB binding protein (Carter and Hunninghake, 2000; Yeh et al., 2004). Therefore, it is reasonable to hypothesize that RAGE-dependent enhancement of MEK–ERK signaling contributes to suppression of NF-κB.
Genetic and Pharmacological Inhibition of RAGE Attenuates LTB4-Dependent Migration of Neutrophils In Vitro and In Vivo
It is well known that LTB4 is a potent lipid chemoattractant for neutrophils, which is mediated by BLT1 (Samuelsson, 1983; Lammermann et al., 2013). To address the role of RAGE in LTB4–BLT1-dependent neutrophil migration, bone marrow-derived neutrophils from wild-type (WT) or RAGE knockout (KO) mice were subjected to chemotaxis assay using LTB4. The velocity of LTB4-dependent neutrophil migration was attenuated in RAGE KO mice compared with WT mice. Furthermore, to determine the pathophysiological role of RAGE, we performed in vivo neutrophil recruitment experiments using an LTB4-induced peritonitis mouse model. The percentage of infiltrating mature neutrophils after LTB4 injection was significantly lower in RAGE KO mice than in WT mice. LTB4-dependent ERK phosphorylation in neutrophils was attenuated in RAGE KO mice, indicating that RAGE potentiates LTB4-dependent neutrophil migration by enhancing ERK phosphorylation.
Previous studies demonstrated that RAGE is also involved in neutrophil chemotaxis, similar to BLT1 (Ryckman et al., 2003; Schiopu and Cotoi, 2013). RAGE binds to various ligands, including cytokine-like S100 proteins, amyloid β, and high-mobility group box 1 (HMGB1) (Yamamoto and Yamamoto, 2012). Interestingly, S100A8 and S100A9, the RAGE ligands, constitute up to 45% of all cytosolic proteins in neutrophils and potentiate neutrophil migration (Ryckman et al., 2003; Schiopu and Cotoi, 2013). These reports support our results, showing that RAGE cooperatively promotes LTB4-dependent neutrophil migration.
RAGE-Dependent Modulation of LTB4–BLT1 Signaling Is a Novel Candidate Molecular Mechanism Underlying Inflammatory Diseases
As already described, we found that RAGE potentiates LTB4-dependent neutrophil recruitment both in vitro and in vivo. Previous studies have reported that the LTB4–BLT1 axis aggravates a variety of acute and chronic inflammatory diseases, all of which are promoted by RAGE. For example, RAGE, with its ligand HMGB1, exacerbates lung injury in mice infected with Staphylococcus aureus pneumonia (Achouiti et al., 2013). RAGE also promotes ovalbumin (OVA)-induced asthma by affecting T cell differentiation (Akirav et al., 2014) and atherogenesis in apoE-deficient mice (Ueno et al., 2010; Yamamoto and Yamamoto, 2013). BLT1 is also involved in protection against infection by bacterial pathogens, including Streptococcus pyogenes (Soares et al., 2013) and Pseudomonas aeruginosa (Lammermann et al., 2013). In addition, BLT1 accelerates OVA-induced asthma by promoting Th2-type immune responses (Terawaki et al., 2005) and enhances atherogenesis in apoE-deficient mice (Heller et al., 2005).
Although the proinflammatory roles of both molecules are quite similar, the functional interplay between BLT1 and RAGE is unknown. Our results provide novel insight into the molecular mechanisms of how acute and chronic inflammatory diseases are regulated. The expression of RAGE is increased by various stimuli, including inflammatory cytokines, advanced glycation end products (Tanaka et al., 2000), high-glucose levels, and reactive oxygen species (Yao and Brownlee, 2010). Thus, environmental inflammatory conditions may activate the LTB4–BLT1 axis through the upregulation of RAGE expression, suggestive of a potential proinflammatory feed-forward system.
RAGE Is a New Class of GPCR Signaling Modifier
We found that RAGE interacts with BLT1. FPR1 and FPRL1 (FPR2/ALX), which are significantly homologous to BLT1, also interact with RAGE (Slowik et al., 2012). The endogenous ligands of BLT1 and FPR2/ALX are lipid mediators, for example, LTB4 for BLT1, and lipoxin A4 and resolvin D1 for FPR2/ALX (Devchand et al., 2003; Krishnamoorthy et al., 2010). This suggests a possible link between RAGE and GPCRs with regard to lipid mediators. Lipid mediators activate many GPCRs, including leukotriene receptors, prostaglandin receptors, free fatty acid receptors, sphingosine 1-phosphate (S1P) receptors, lysophosphatidic acid (LPA) receptors, and proresolving lipid mediator receptors in vertebrates (Hla et al., 2008; Blaho and Hla, 2014; Yung et al., 2014; Chiang et al., 2015). In the same way as RAGE and BLT1, the S1P and LPA receptor signaling systems contribute to immune cell trafficking. The vascular S1P gradient is required for lymphocyte trafficking through the chemotactic activity of S1P1 (Hla et al., 2008; Blaho and Hla, 2014). LPA also regulates immune cell trafficking through multiple LPA receptors (Yung et al., 2014).
Of the GPCRs bound by lipid mediators, BLT2 is of particular interest. BLT2 was first cloned as a low-affinity receptor for LTB4 (Yokomizo et al., 2000a) but is now considered a high-affinity receptor for the oxidized fatty acid 12(S)-hydroxyheptadeca-5Z, 8E, 10E-trienoic acid (12-HHT), and its functions are not yet known (Okuno et al., 2008). In mice, BLT2 is expressed in intestinal epithelial cells and epidermal keratinocytes (Iizuka et al., 2005), whereas human BLT2 is ubiquitously expressed (Yokomizo et al., 2000b). The 12-HHT–BLT2 axis plays important roles in intestinal and skin barrier function (Iizuka et al., 2010; Ishii et al., 2016) and skin wound healing through accelerating keratinocyte migration (Liu et al., 2014). In addition to the fact that BLT2 is highly homologous to human and mouse BLT1, with amino acid identities of 45.2% and 44.6%, respectively, the tissue distribution of BLT2 is similar to that of RAGE, with high expression in keratinocytes (Sakaguchi et al., 2014). Future studies will focus on the potential physical and functional interactions between RAGE and other GPCRs with lipid mediators, including BLT2.
Concluding Remarks
We identified RAGE as a novel binding protein for BLT1 and showed that RAGE modulates downstream signaling from BLT1 and affects cell migration through BLT1 both in vitro and in vivo (Fig. 1). Our findings provide insight into the roles of RAGE in the LTB4–BLT1 signaling pathway and contribute to our understanding of the role of RAGE as a GPCR signaling modifier.
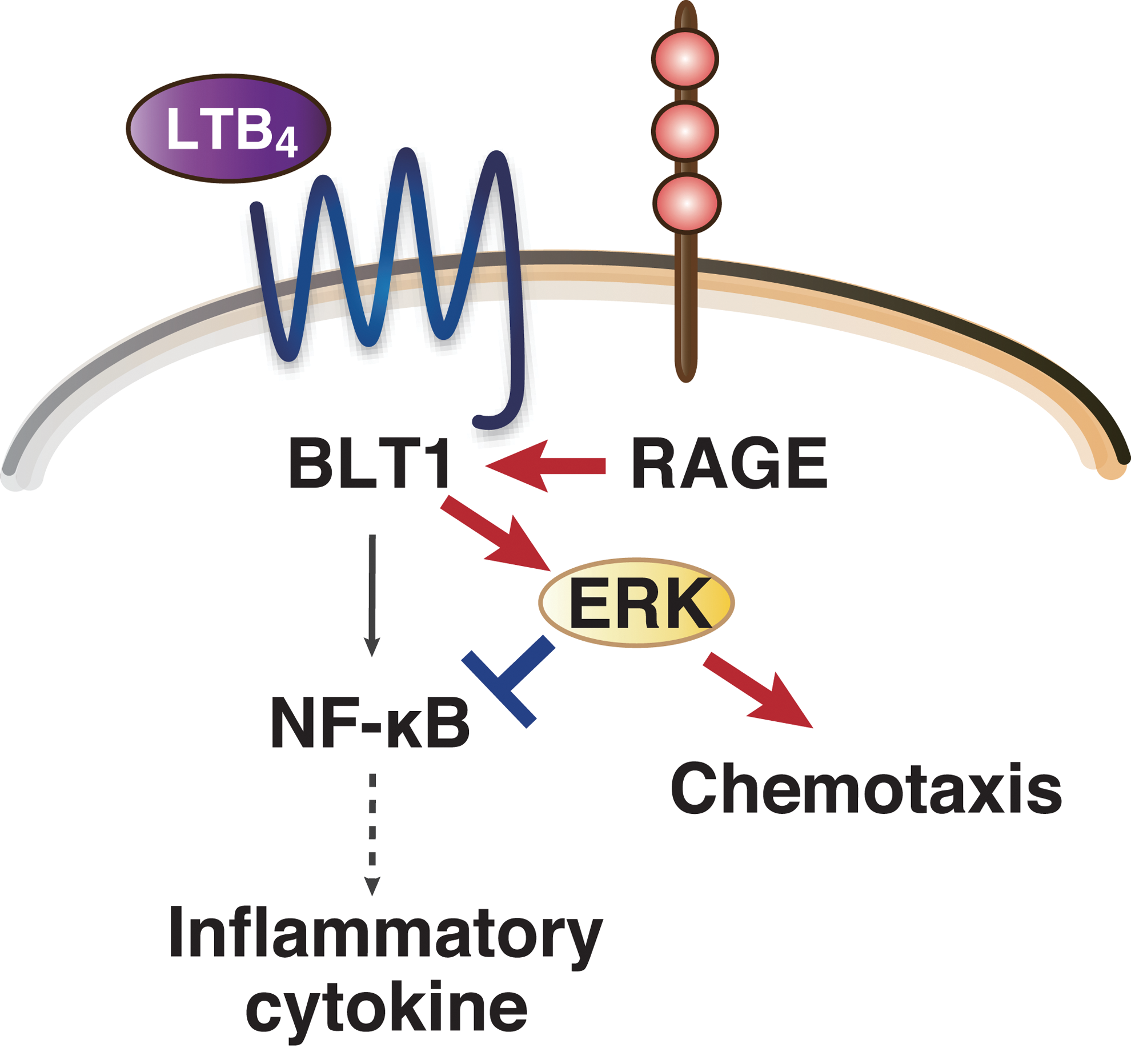
The BLT1–RAGE interaction modulates LTB4–BLT1 signaling both positively and negatively. RAGE interacts with BLT1 and enhances LTB4–BLT1-dependent ERK phosphorylation. Whereas RAGE potentiates LTB4-dependent neutrophil chemotaxis, RAGE suppresses LTB4–BLT1-dependent NF-κB activation and upregulation of inflammatory cytokines. Both positive and negative regulation of LTB4–BLT1-dependent output by RAGE is mediated by potentiation of ERK phosphorylation. BLT1, leukotriene B4 receptor 1; ERK, extracellular signal-regulated kinase; LTB4, leukotriene B4; RAGE, receptor for advanced glycation end products.
Footnotes
Acknowledgments
This work was supported by MEXT/JSPS KAKENHI Grant numbers, 22116001, 22116002, 15H05901, 15H05904, 15H04708, 15K08316, 15K19032, 22860223, 24590386, and 25460374, and Grants from the Naito Foundation, the Ono Medical Research Foundation, the Uehara Memorial Foundation, the Mitsubishi Foundation, the Nakatomi Foundation, and the Takeda Science Foundation. This study was supported, in part, by a Grant-in-Aid (S1311011) from the Foundation of Strategic Research Projects in Private Universities from the MEXT and a Grant of Institute for Environmental and Gender-Specific Medicine.
Disclosure Statement
No competing financial interests exist.