
Abstract
A-to-I RNA editing, carried out by adenosine deaminase acting on RNA (ADAR) enzymes, is an epigenetic phenomenon of posttranscriptional modifications on pre-mRNA. RNA editing in intronic sequences may influence alternative splicing of flanking exons. We have previously shown that conditions that induce editing result in elevated expression of signal transducer and activator of transcription 3 (STAT3), preferentially the alternatively-spliced STAT3β isoform. Mechanisms regulating alternative splicing of STAT3 have not been elucidated. STAT3 undergoes A-to-I RNA editing in an intron residing in proximity to the alternatively spliced exon. We hypothesized that RNA editing plays a role in regulating alternative splicing toward STAT3β. In this study we extend our observation connecting RNA editing to the preferential induction of STAT3β expression. We study the involvement of ADAR1 in STAT3 editing and reveal the connection between editing and alternative splicing of STAT3. Deferoaxamine treatment caused the induction in STAT3 RNA editing and STAT3β expression. Silencing ADAR1 caused a decrease in STAT3 editing and expression with a preferential decrease in STAT3β. Cells transfected with a mutated minigene showed preferential splicing toward the STAT3β transcript. Editing in the STAT3 intron is performed by ADAR1 and affects STAT3 alternative splicing. These results suggest that RNA editing is one of the molecular mechanisms regulating the expression of STAT3β.
Introduction
P
In primates, most of the edited sites reside in noncoding regions, typically in Alu elements. The pairing of adjacent inverted Alu elements leads to the formation of extended base-paired duplex dsRNA resulting in a secondary structure, which may undergo extensive editing by the adenosine deaminase acting on RNA (ADAR) editing enzymes. In humans ADAR1 (isoforms, p110 and p150) and ADAR2 are responsible for all currently known A-to-I editing activity. These modifier enzymes have distinct but overlapping specificities. A-to-I editing events can be identified as sites at which a guanosine (G) has replaced an adenosine (A) since the translational and splicing machineries recognize inosine as guanosine (Daniel et al., 2015).
Recent studies revealed that nearly all multiexon human genes (comprising >90% of all genes) generate alternative mRNA isoforms, and most do so in a tissue specific manner although the mechanisms leading to the alternative transcripts are not always known (Pan et al., 2008). A-to-I RNA editing has been linked to alternative splicing in several cases. ADAR2 autoregulates its own expression by alternative splicing. ADAR2 edits its own pre-mRNA and creates an alternative 3′ splice site. The alternative spliced isoform contains a premature stop codon resulting in a dysfunctional truncated protein (Rueter et al., 1999).
In another case, hyper editing in the human NARF gene creates a 3′ splice-site consensus sequence, which leads to exonization of an intronic sequence and the birth of a new exon (Sorek et al., 2002; Lev-Maor et al., 2003, 2007). Furthermore, in vitro experiments in the human RABL5 gene demonstrated that de novo insertion of an Alu element in the antisense orientation to the adjacent Alu element within an intron causes the downstream exon to shift from constitutive splicing to alternative splicing, as seen in Glycerol kinase deficiency and Apert syndrome (Oldridge et al., 1999; Zhang et al., 2000; Lev-Maor et al., 2008).
The signal transducer and activator of transcription 3 (STAT3) gene is a member of the STAT family of transcription factors. It has been associated with a wide variety of normal and pathological cell states. The gene encodes two main isoforms, which are translated into two distinct proteins: STAT3α (p92) and STAT3β (p83) (Qi and Yang, 2014). These isoforms are generated by alternative splicing: the short form (STAT3β) is generated by an alternative 3′ acceptor site 50 nucleotides within exon 23 and lacks 55 C-terminal amino acid residues of the long form (STAT3α), including the transactivator domain (Fig. 1A, B). Seven additional amino acid residues are encoded at its C terminus by 21 nucleotides spliced in the +2 reading frame (Qi and Yang, 2014). To date, the full molecular mechanisms regulating the alternative splicing of STAT3 have not been elucidated.
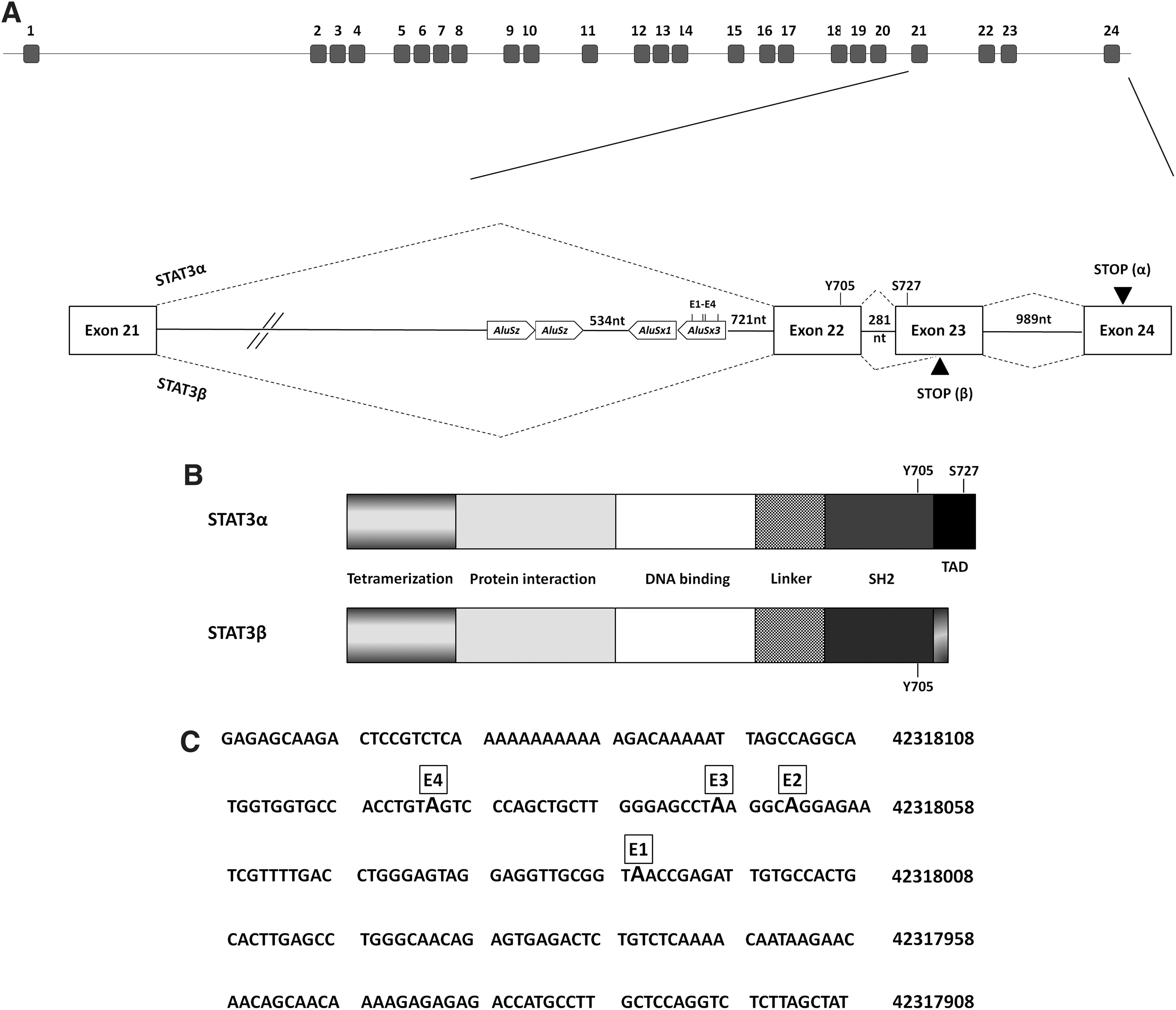
STAT3 gene, transcript and protein organization.
STAT3 has been implicated in growth factor and cytokine signaling. Its activation involves phosphorylation at tyrosine705 and Serine727. Phosphorylation of STAT3 Tyr705 is critical for STAT3 dimerization and nuclear translocation, whereas the phosphorylation of Ser727 enhances STAT3 transcriptional activity. The phosphorylated STAT3 dimerizes, migrates to the nucleus, and binds to cognate elements on the promoters of responsive genes (Levy and Lee, 2002; Liu et al., 2005).
Since STAT3β does not have a transactivation domain and lacks Serine,727 it was initially thought to be ineffective in activating gene transcription and believed to act as a dominant negative reducing STAT3α function (Caldenhoven et al., 1996). However, additional studies revealed a role for STAT3β in gene transcription (Maritano et al., 2004; Zammarchi et al., 2011; Marino et al., 2014); mice lacking STAT3β were more sensitive to endotoxic shock, exhibiting increased mortality and tissue damage after exposure to LPS (Lipopolysaccharide) (Yoo et al., 2002).
We have previously shown that a human lymphoblastoid (LB) cell line exposed to deferoaxamine (DFO), which mimics hypoxic conditions (Ivan et al., 2001; Zeng et al., 2011), exhibited increased expression of the ADAR editing enzymes followed by elevated levels of A-to-I RNA editing in an intronic Alu element embedded in a STAT3 intron, ∼1 kb upstream to the alternatively spliced exon 23 (Fig. 1) (Nevo-Caspi et al., 2011). In addition we demonstrated that DFO-treated cells show elevated expression levels of STAT3 with a preferential induction of the alternatively spliced STAT3β isoform (Nevo-Caspi et al., 2011).
In the current study we extend these observations to the MCF7 cell line and we examined if RNA editing in STAT3 affects the splicing pattern of the gene. We found that upon silencing of ADAR1, STAT3 editing is reduced with a concomitant decrease in STAT3 expression, mainly the STAT3β isoform. Cloning of a STAT3 minigene enabled us to simulate 100% editing efficiency at STAT3 and we show that this change affected the splicing pattern of the alternatively spliced flanking exon. As a result, higher levels of STAT3β were transcribed. We therefore conclude that RNA editing influences STAT3 alternative splicing. We suggest that this is one of the molecular mechanisms used to regulate STAT3 expression.
Materials and Methods
Cell lines
A human LB cell line was generated by Epstein–Barr virus-transformation of B cells originating from a healthy male donor. Ethics statement: The Institutional Review Board (Sheba Medical Center, Tel Hashomer) approved human involvement and cell line creation in this study. Written consent to participate in this study was obtained from donor. MCF7 cells originate from a human invasive breast ductal carcinoma and were obtained from American Type Culture Collection (ATCC). LB cells were cultured in RPMI and MCF7 cells were cultured in DMEM, both supplemented with 10% fetal bovine serum, 1% penicillin:streptomycin, and 1% glutamine, at 37°C in a humidified incubator with 5% CO2.
DFO treatment
To mimic hypoxia, cells were treated for 24 h with 1000 μM Deferoaxamine (Sigma-Aldrich). Cell viability will not be affected under these conditions.
siRNA
The siRNAs used were Ambion Silencer Validated siRNAs: Negative control cat. no. AM4635, ADAR1 cat. no. 119581, and ADAR2 cat. no. 119782. The optimal concentrations of siRNA were 20 μM for ADAR1 and 40 μM for ADAR2.
Transfection of siRNA
LB cells (4 × 106 per well) were pelleted and resuspended in 90 μL of Ingenio Electroporation Solution (Mirus) containing the relevant siRNA. Each sample was electroporated in the Nucleofector Device (Amaxa Biosystems) using the R-13 program. Cells were transferred into 2 mL prewarmed medium and cultured for a period of 24 h; subsequently the expression of mRNA and proteins was analyzed. All experiments were repeated at least thrice.
MCF7 cells (2.5 × 105 cells per well) were transfected using Lipofectamine® 2000 (Life Technologies). Two hundred microliters of siRNA complexes in 1 mL fresh medium were added, and the cells were incubated at 37°C for 4 h followed by the addition of 1 mL medium. After 24 h, the expression of mRNA and proteins was analyzed. All experiments were repeated at least thrice.
Transfection of plasmids
LB cells (2.5 × 106 per well) were pelleted and resuspended in 90 μL of BTXpress electroporation buffer (Harvard BTX) and 2 μg of plasmid in 10 μL H2O. Each sample was electroporated using 0.2 cm cuvettes in the ECM 830 Square Wave Electroporation System (Harvard BTX). The following electroporation settings were used: voltage: 100 V; pulse length: 65 ms; and number of pulses: 1.
Following electroporation the cells were transferred from the cuvettes into six-well plates containing 2 mL prewarmed RPMI medium. Cells were cultured for a period of 24 h; subsequently the expression of mRNA was analyzed.
RNA extraction and cDNA preparation
Total RNA was extracted from cells using the TRIzol reagent (Invitrogen) according to the manufacture's protocol. cDNA was prepared from 2 μg of total RNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacture's protocol.
Polymerase chain reaction amplification and sequence reactions
Polymerase chain reaction (PCR) amplifications were performed using ReddyMix PCR Master Mix (ABgene). For sequencing reactions, PCR fragments were amplified with the following primers (5′ to 3′): STAT3-F: CATAAAAGAGAATATGGGCTGGGC; STAT3-R: GGTCCAGGTACATCTTCAATAGCT. Amplified fragments were purified from gel using Zymoclean Gel DNA Recovery Kit (Zymo Research) according to the manufacture's protocol. Purified DNA was fluorescently labeled using the BigDye Terminator V1.1 cycle enzyme (Applied Biosystems) with the STAT3-R2 primer (see above) according to the manufacture's protocol. Following precipitation, fragments were sequenced using the ABI PRISM 3100 Genetic Analyzer Biosystem.
Analysis of A-to-I RNA editing
Editing in STAT3 was examined in an Alu sequence embedded in the 21st intron of the STAT3 gene positioned at chr17:42317890-42318460 according to the UCSC genome assembly hg38. cDNA extracted from the cells was amplified using the following primers:
F-5′CATAAAAGAGAATATGGGCTGGGC3′
R-5′GGTCCAGGTACATCTTCAATAGCT3′
Inosine is similar in structure to guanine and is, therefore, read by the automatic DNA sequencer as “G.” A-to-I RNA editing is identified as a site in the cDNA sequence with overlapping A and G peaks. The percentage of RNA editing was measured using the Accelrys Gene software by calculating the level of guanine expression at a certain sequence position divided by the total expression of adenine and guanine at the same point. Edited sites are only those sites that exhibited more than 5% editing. Positions of the editing sites, according to UCSC genome assembly hg38, are as follows: E1—42318026; E2—42318064; E3—42318069; and E4—42318091.
Relative quantification analysis
Relative quantification (RQ)-PCR was performed to determine the levels of mRNA expression of ABL, ADAR1 (p110 and p150), ADAR2, and STAT3 (α and β). We used the ABL gene as a reference gene, which showed stable levels of expression in our treated and nontreated samples (data not shown). Primers and probes were designed according to Primer Express software guidelines (Applied Biosystems) to enable the amplification of mature mRNA only (Table 1). All probes carried a 5′FAM and a 3′BHQ. RQ-PCR was run on ABI 7900HT Genetic Analyzer utilizing SDS 2.3 software (Applied Biosystems). All reactions were run as triplicates. Reactions were performed in a total volume of 20 μL containing cDNA equivalent to 100 ng of RNA from each sample. The PCR conditions consisted of 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. Analysis of the results was performed by the SDS RQ manager 1.2 software using the ΔΔCt method.
ADAR, adenosine deaminase acting on RNA; STAT3, signal transducer and activator of transcription.
Western blot analysis and antibodies
Proteins were extracted from cells using RIPA buffer (Sigma-Aldrich) supplemented with protease inhibitor (Roche). Following separation on a SDS-PAGE, proteins were transferred to a nitrocellulose membrane followed by staining with a primary antibody overnight at 4°C and washed and incubated with the appropriate secondary antibody for 1 h at room temperature. Specific reactive bands were detected using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). RQ of the proteins was performed using the ImageJ software. The antibodies used were as follows: anti-ADAR1 (Santa Cruz sc-73408), anti-STAT3 (Cell Signaling no. 9139), anti-actin (Abcam Ab156302), and anti-tubulin (Abcam T9026).
STAT3 minigene construction
Human genomic DNA was amplified by PCR with the Platinum Pfx enzyme (Invitrogen) following manufacturer's instructions for 35 cycles, T m 60°C, and a 5 min extension time, using the following primers:
5′ATATATGGATCCTCAGGACGATGAGCATGTGACCA3′
5′TAATAAGCGGCCGCGCTTAAAGCACCAAGGAGGCTGT3′
The 5.0 kb amplified fragment was digested with BamHI and NotI and cloned into pcDNA3 linearized with BamHI and NotI. The resulting plasmid was sequenced.
STAT3 mutated plasmid construction
Site directed mutagenesis was performed with the QuikChange II Site-Directed Mutagenesis Kit according to the manufacture's protocol (Stratagene). Briefly, specific overlapping primers (Table 2) containing the desired mutations at the four edited sites (E1–E4) were used to amplify a mutation-containing replica of the wild-type minigene plasmid. Following PCR amplification the products were treated with DpnI restriction enzyme at 37°C for 1 h. The mutant DNA (5–10 μL) was transformed into MAX Efficiency® Escherichia coli DH5α-competent cells according to the manufacture's protocol (Invitrogen). Plasmid extraction was performed from colonies and screened by restriction enzyme digestion. All mutated plasmids were confirmed by sequencing.
Statistical analysis
The T-test was used to calculate statistical differences between two samples. Results are given as mean value ± SD. In graphs, columns marked with asterisks are significantly different than the control sample (p-value <0.01).
Results
We added another cell line to our study to provide greater insight into the effect of DFO on STAT3 editing and STAT3β expression. MCF7 cells, which originated from a human invasive breast carcinoma, were treated with DFO, after which A-to-I RNA editing in the STAT3 gene was studied. Of the several edited sites that were detected, we focused on the four (E1–E4) that showed more than a 5% change in the amount of edited molecules upon DFO treatment (Fig. 2A) (the editing levels at sites E5–E12 are shown in Supplementary Fig. S1; Supplementary Data are available online at
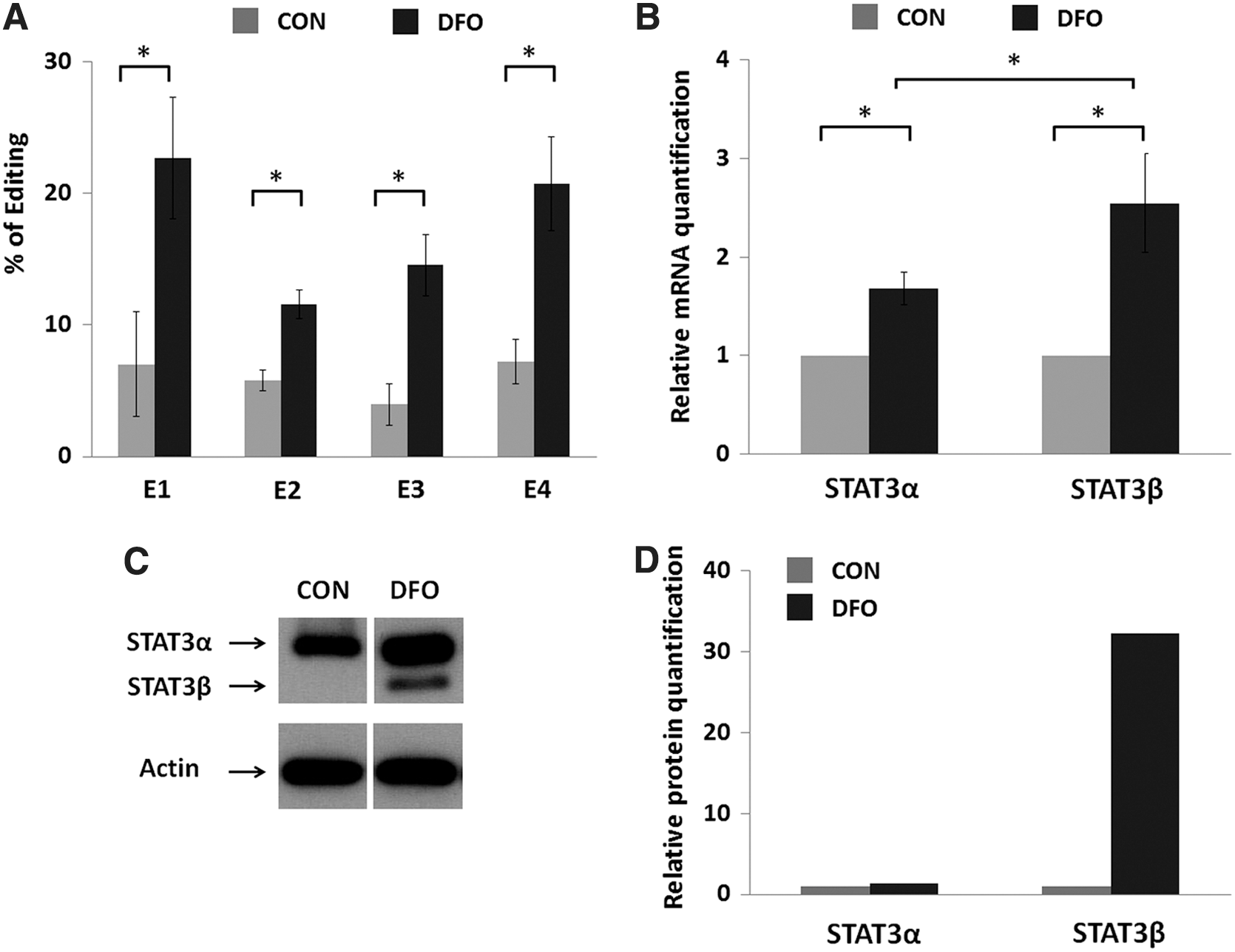
A-to-I RNA editing and mRNA and protein expression in MCF7 cells upon DFO treatment.
RQ analysis showed that following DFO treatment the expression of both STAT3 isoforms was elevated. STAT3β expression was elevated to a larger extent (Fig. 2B). A similar result was obtained at the protein level. The amount of the STAT3α protein was greater following DFO treatment, and the STAT3β protein was detected only in the DFO-treated sample (Fig. 2C, D). We therefore conclude that the phenomenon of DFO-induced RNA editing and induced STAT3 expression (preferentially STAT3β) is not restricted only to the LB cell line and is probably a broad one.
The edited Alu in the STAT3 gene can form a dsRNA
To investigate if a double-stranded region can be formed between the STAT3 intronic Alu element that we have previously shown to undergo editing and a complementary sequence (probably one of the two inverted Alu intronic elements located upstream, see Fig. 1A), we used the RNA folding and hybridization software Mfold (Zuker, 2003). Indeed this analysis revealed that the edited AluSx3 element can form a long dsRNA structure with an AluSz sequence situated in the same intron, ∼1.1 kb upstream (Fig. 3). This double-stranded region is adjacent to an alternatively spliced STAT3 exon. We therefore hypothesized that editing in the Alu sequence may affect the alternative splicing of the gene.
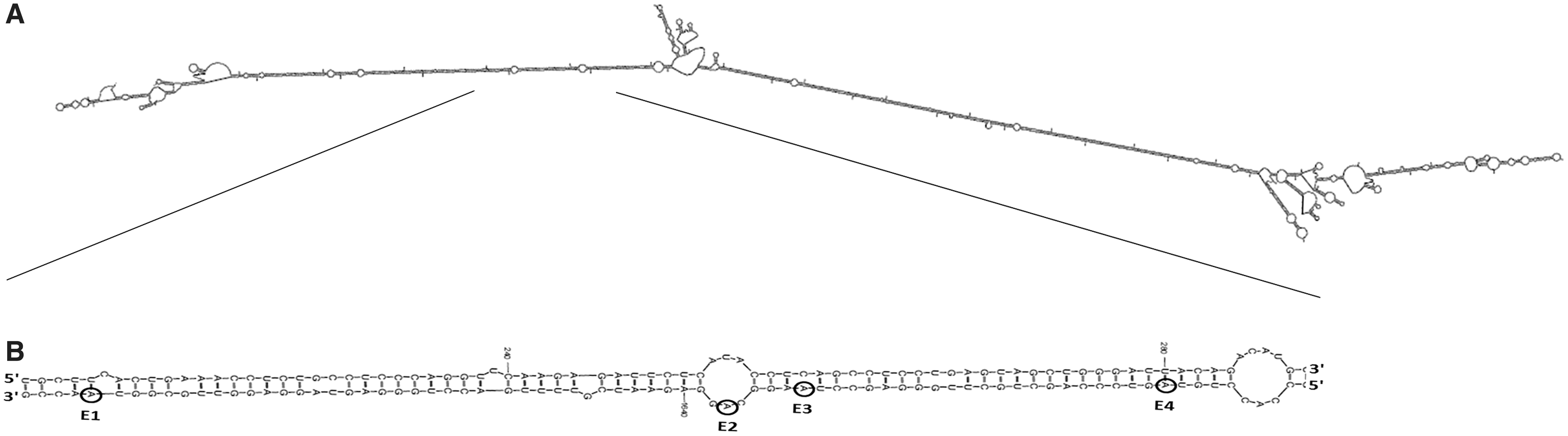
Predicted pre-mRNA secondary structure according to the Mfold algorithm (Zuker, 2003).
ADAR1 is responsible for the editing in the STAT3 gene
ADAR1 and ADAR2 are both known to catalyze A-to-I RNA editing in mammals. We attempted to determine which of these enzymes was responsible for the editing events seen in STAT3. To address this question we silenced ADAR1 or ADAR2 expression by transfecting the LB cells with specific siRNAs. As a control the cells were transfected with a nontargeting siRNA. RNA was extracted from the cells 24 h following transfection and subjected to RQ-PCR and editing analysis. Proteins extracted from the transfected cells were analyzed by Western blot.
LB cells transfected with a specific siRNA targeted toward a common region in the transcripts of ADAR1 showed a decrease in mRNA and protein levels of both ADAR1 subunits, p110 and p150 (Fig. 4A–C). ADAR2 levels remained unchanged in these experiments. Indeed, sequencing of RT-PCR products of the edited region of STAT3 showed decreased editing levels at the four edited sites (E1–E4) implying that ADAR1 is responsible for these events (Fig. 4D). Analysis of STAT3 expression by RQ-PCR and Western blot revealed that in the ADAR1-silenced cells, the expression of STAT3 was reduced. A more pronounced reduction was seen for STAT3β (Fig. 4A–C). These results suggest an association between the editing in the STAT3 intron and the splicing toward the STAT3β isoform. Silencing of ADAR2 did not reveal changes in editing levels in STAT3 or in the levels of STAT3 RNA (Supplementary Fig. S2).
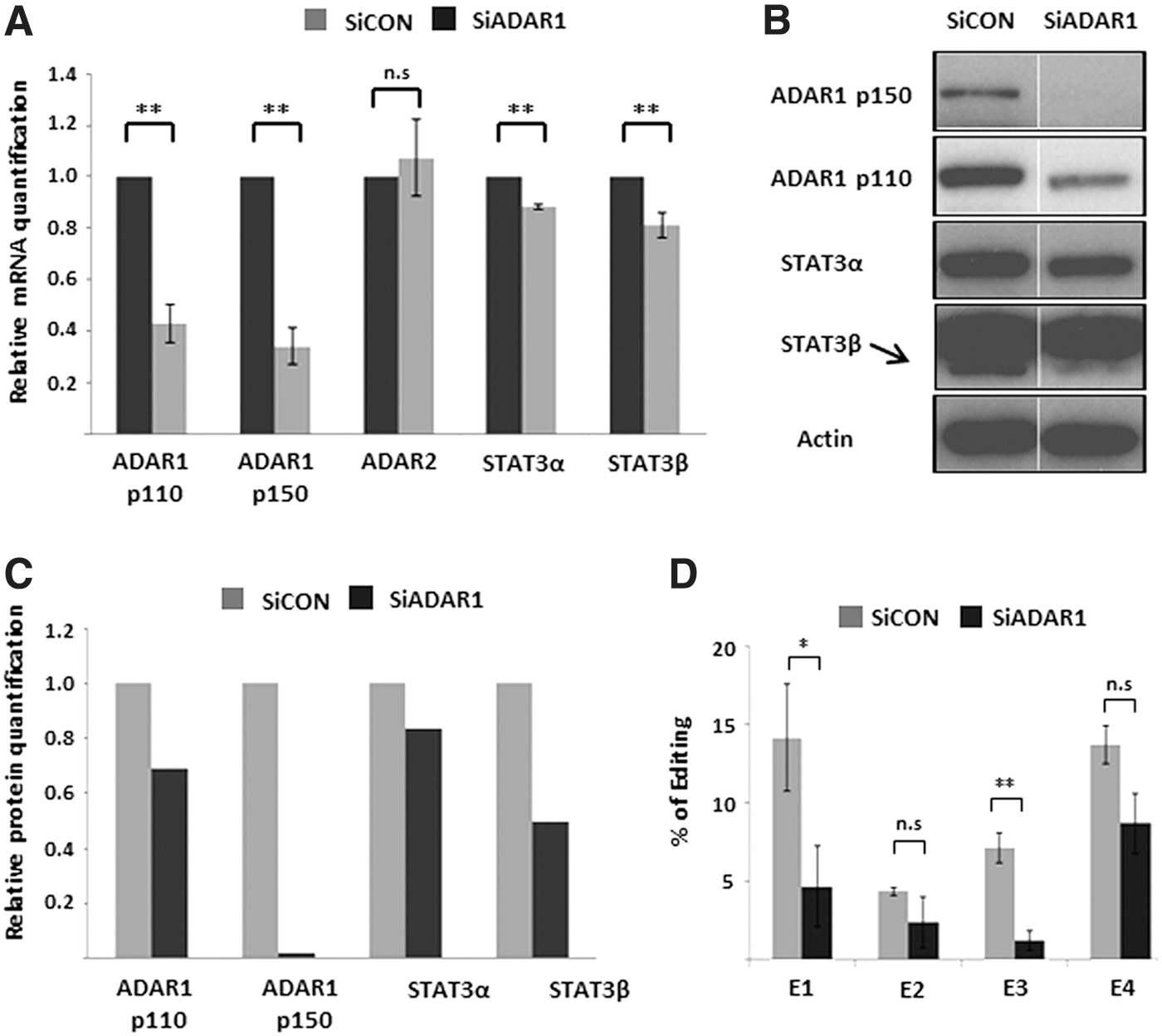
mRNA, protein expression, and A-to-I RNA editing upon silencing of ADAR1 in LB cells.
Similar silencing experiments and analyses were performed in the MCF7 cells. Results regarding the involvement of ADAR1 in STAT3 editing and the effect of ADAR1 silencing on the decreased STAT3β expression were similar to those obtained in LB cells (Fig. 5A–D).
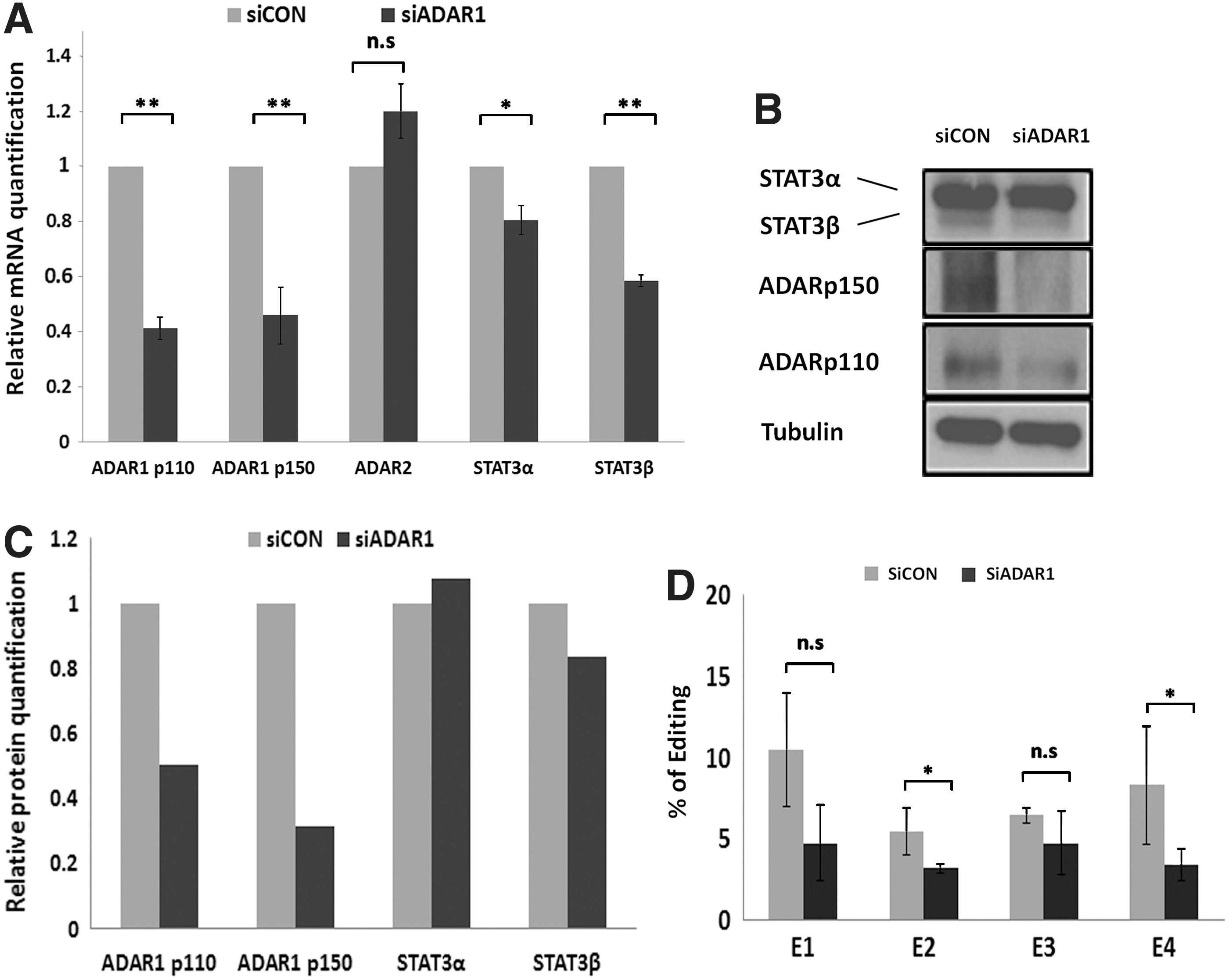
mRNA, protein expression, and A-to-I RNA editing upon silencing of ADAR1 in MCF7 cells.
RNA editing affects STAT3 alternative splicing
To further study a possible association between A-to-I RNA editing in the STAT3 intron and the splicing toward the STAT3β isoform, we cloned a minigene bearing the genomic region of STAT3 spanning intron 21 (including the edited sites) through exon 24 (Fig. 1). We then mutated each of the four intronic E1–E4 sites in the plasmid from A-to-G, mimicking 100% editing efficiency at these sites.
LB cells were transfected with the STAT3-mutated plasmids and as a control with the STAT3 wild-type plasmid. Twenty-four hours posttransfection cells transfected with the mutated (edited) plasmid showed a significant 1.7-fold increase in STAT3β transcript levels compared to the amount obtained with the WT plasmid (Fig. 6). No difference was obtained in the amount of the STAT3α transcripts. We therefore conclude that “edited” plasmids were favorably spliced into the STAT3β isoform implying that A-to-I RNA editing of the STAT3 intronic AluSx3 affects the splicing pattern of the alternative spliced flanking exon. As a result, higher levels of the STAT3β transcript are obtained.
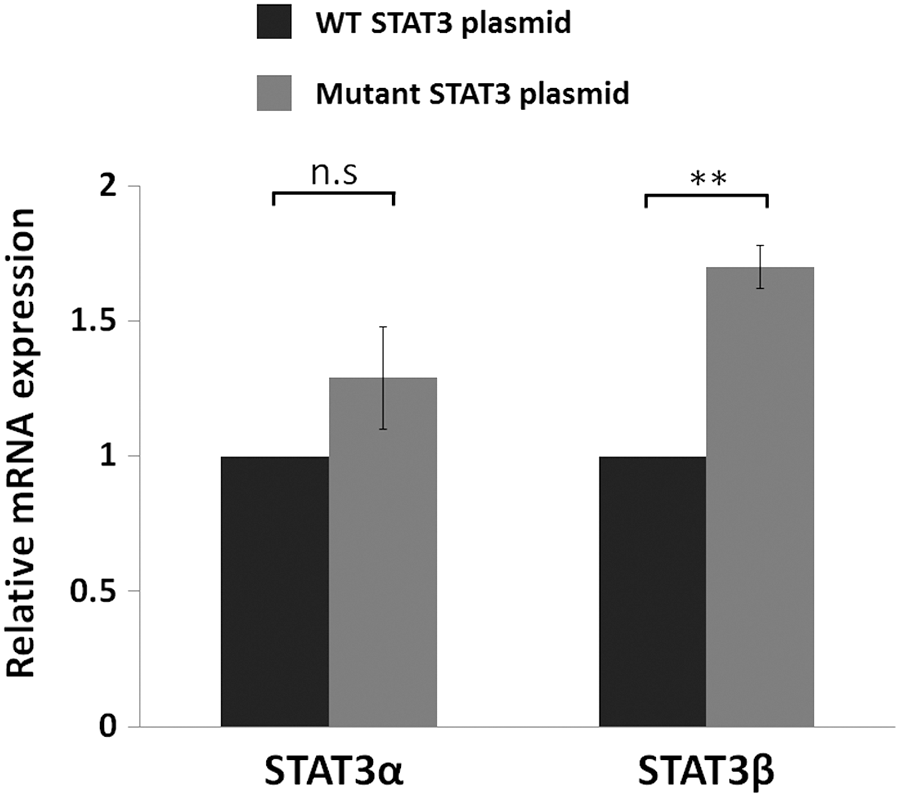
mRNA expression of STAT3 upon transfection with the STAT3 plasmid. STAT3 RNA expression levels of LB cells transfected with the WT STAT3 plasmid or the mutated STAT3 plasmid were evaluated by RQ-PCR. Results were normalized to the RNA levels obtained with the transfection of the WT plasmid which was set as 1. Results are the mean of three repeats of each experiment ± SEM. **p ≤ 0.01.
Discussion
In this study we have extended to an additional cell line, our previous observation that DFO treatment causes an induction in STAT3 RNA editing and in STAT3 expression, preferentially the alternatively spliced isoform—STAT3β. We therefore conclude that this phenomenon is a broad one emphasizing its importance in regulating STAT3 levels in the cell. In addition, we show that ADAR1 is responsible for these editing events and that A-to-I RNA editing that occurs in an intronic Alu of the STAT3 gene affects the alternative splicing that occurs in the adjacent exon 23, encouraging the formation of STAT3β, a shorter isoform of the gene.
STAT3 modulates the transcription of a variety of genes involved in the regulation of critical functions, including cell differentiation, proliferation, apoptosis, angiogenesis, metastasis, and immune response (Aaronson and Horvath, 2002; Levy and Lee, 2002; Aggarwal et al., 2009). As such, STAT3 expression is expected to respond to many different environmental cues. Indeed, STAT3 has been reported to stimulate the production of inflammatory factors upon cancer and to respond to the hypoxic microenvironment in a variety of tumors (Yu et al., 2009).
The STAT3 gene encodes two main isoforms as follows: STAT3α and truncated STAT3β generated by alternative splicing in exon 23. The two proteins translated from these isoforms have been shown to play distinct roles in transcriptional regulation (Huang et al., 2007). Specifically, distinctive STAT3β properties and activities have recently been described, indicating that STAT3β is not simply a truncated version of STAT3α. Phosphorylation of STAT3 Tyr705 is critical for dimerization, nuclear import, and transcriptional activity. Under the same cytokine stimulation conditions, STAT3β showed remarkable prolonged phosphorylation and nuclear retention compared with STAT3α. Furthermore, STAT3β enhanced and prolonged the phosphorylation and nuclear retention of STAT3α (Ng et al., 2012).
Transcriptome profiling of STAT3−/− cells, expressing either STAT3α or STAT3β, revealed a distinct set of STAT3β-specific genes regulated under basal conditions and following cytokine stimulation (Yoo et al., 2002; Maritano et al., 2004). These results highlight the role of STAT3β as a potent transcriptional regulator and its ability to cross-regulate and enhance the transcriptional activity of STAT3α. In vivo, STAT3β-deficient mice exhibit diminished recovery from endotoxic shock and upregulation of a subset of inflammation-related genes (Yoo et al., 2002). In apolipoprotein E-deficient mice, selective removal of STAT3β exacerbates the development of atherosclerosis, indicating that STAT3β can suppress pathologic consequences associated with chronic inflammation (Lee et al., 2013). Moreover, STAT3β expression rescued embryonic lethality in STAT3−/− mice (Maritano et al., 2004).
Oligonucleotide-mediated enforced switching to preferential splicing of STAT3β was found to be in association with increased cell death in cancer cells and with tumor regression in a xenograft model (Zammarchi et al., 2011). The different transcriptional roles attributed to both STAT3 isoforms suggest that the alternative splicing of this gene must be carefully regulated. However, to date, the full molecular mechanisms regulating this event have not been elucidated. In this study, we show that RNA editing in humans is associated with this splicing event. We show that A-to-I RNA editing that occurs in a STAT3 intronic Alu leads to an induction in STAT3β levels suggesting that conditions leading to induced RNA editing, such as hypoxia, will result in higher STAT3β levels (Nevo-Caspi et al., 2011).
Emerging evidence suggests that the pre-mRNA secondary structure plays a role in alternative splicing (Morse et al., 2002). The secondary structures can inhibit or activate spliceosome assembly by affecting the recognition of splice sites, enhancers, and silencers, which depend upon interactions between splicing regulatory proteins and a single-stranded portion of the pre-mRNA (Shepard and Hertel, 2008). For example, in the human survival motor neuron (SMN) gene, skipping of exon 7 is regulated by the RNA secondary structure, which influences a 5′ splice site (Singh et al., 2007). In the fibroblast growth factor receptor 2 (FGFR2) gene dsRNA stem structure regulates the splicing of mutually exclusive exons (Muh et al., 2002). Bioinformatic analysis supports the notion that RNA secondary structures modulate pre-mRNA splicing in humans and suggests that stable and long-range RNA secondary structures are associated with alternative splicing (Shepard and Hertel, 2008; Pervouchine et al., 2012; Solomon et al., 2013).
Our hypothesis that the region of the edited Alu element may be involved in a secondary RNA structure that may affect the alternative splicing in STAT3 exon 23 was enforced by our analysis using the Mfold software that revealed a long secondary structure, which can form between the AluSx3 element and the adjacent AluSz situated ∼1.1 kb upstream (Fig. 3). As mentioned above, alternative splicing of STAT3 leading to the formation of STAT3β occurs in mice as well. Although repetitive elements are present in the mouse relevant intron, no editing events are predicted at the site (Neeman et al., 2006; Ramaswami and Li, 2014). Several studies have found that the editing levels are lower in mice than in humans (Eisenberg et al., 2005; Neeman et al., 2006). We therefore believe that splicing toward STAT3β in mice is not affected by intronic A-to-I RNA editing.
RNA editing has been previously linked to alternative splicing in several cases. In the ADAR2 and the glutamate receptor GRIA2 (formerly GluR-B) genes, RNA polymerase II C-terminal domain is thought to coordinate RNA processing events, including editing and splicing (Laurencikiene et al., 2006; Ryman et al., 2007). The most notable example for demonstrating coupling between editing and alternative splicing is the autoediting of ADAR2, which modulates its own expression (Rueter et al., 1999). A-to-I RNA editing occurring in Alu elements dispersed throughout the human genome has similarly been linked to alternative splicing (Bentley, 2014). Our study provides additional insights for the close relationship between alternative splicing and remote A-to-I RNA editing and their contribution to transcriptome diversity.
Additional methods for identifying new potential editing sites may reveal the existence of additional sites that have the ability of affecting splicing toward the STAT3β isoform. We show that even editing solely at sites E1–E4 caused a change in the alternative splicing toward STAT3β. To the best of our knowledge, this is the first study that attributes a molecular mechanism to the splicing event leading to the formation of the STAT3β isoform. In our study, silencing of ADAR1 caused a decrease in the expression of STAT3β. We note, however, that in addition to the decrease in STAT3β, a slight decrease in STAT3α was seen. Changes in the levels of STAT3α may result from additional roles attributed to ADAR1.
Editing-independent mechanisms have been suggested to be responsible for global changes in the cellular splicing pattern. Solomon et al. (2013) reported in their study that the small number of edited splicing regulatory elements that they detected could not explain the massive changes observed in splicing upon ADAR1 knockdown, and therefore, they suggested that a significant part of the ADAR-dependent alternative splicing regulation is not a result of direct editing activity, but is mediated by additional factors, including editing-independent mechanisms. As part of their study, Solomon et al. indeed observed changes in STAT3 alternative splicing following ADAR1 knockdown; however, their analysis did not distinguish between the two STAT3 isoforms.
Our results with the mutated STAT3 plasmid (namely changes in STAT3β expression with no change in STAT3α expression) indicate that A-to-I RNA editing does play a role in the alternative splicing event, which results in the formation of the STAT3β isoform and support the idea that the slight changes obtained in the expression of STAT3α upon changes in ADAR1 expression are due to editing-independent ADAR1 effects, which remain to be studied. In accordance with our results, Wang et al. (2013) found editing differences in the AluSx3 element embedded in the STAT3 intron in sequences obtained from B cells from two individuals. They next identified one of these sites (E1 site in our study) in an anti-ADAR1 RNA-immunoprecipitation experiment implicating that this site indeed undergoes ADAR1-dependent RNA editing.
Several studies have documented the close relationship between STAT3 and hypoxia: STAT3 and HIF1α, known as the master transcriptional regulator of the cellular response to hypoxia, have been shown by several groups to cooperate (Jung et al., 2005; Niu et al., 2008; Pawlus et al., 2014); in glioblastoma cells hypoxic conditions increased phosphorylated (activated) STAT3 levels and STAT3-dependent angiogenesis and cell migration (Kang et al., 2010).
In addition to this study and a previous one from our laboratory (Nevo-Caspi et al., 2011), additional groups have now documented the induction of RNA editing upon hypoxic cell conditions (Nigita et al., 2016). Moreover, we have previously documented elevated levels of A-to-I RNA editing in cyanotic pediatric patients (Borik et al., 2011). Following our results in the present study we conclude that the DFO-dependent elevated levels of STAT3β were a result of the induced editing, whereas the slight induction in STAT3α levels resulted from an editing-independent action of ADAR1.
In conclusion, we have shown that ADAR1-dependent A-to-I RNA editing in the STAT3 intronic Alu influences the alternative splicing of the gene. These results interconnect the hypoxia-induced A-to-I RNA editing and the preferential induction of alternatively spliced STAT3β seen under hypoxic conditions. We suggest that this is one of the molecular mechanisms used in response to hypoxic stress.
Footnotes
Acknowledgment
The authors thank Amisragas for their continuous support of the Department of Intensive Care.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.