
Abstract
Phytoplasmas are obligate intracellular parasitic bacteria that infect both plants and insects. We previously identified the sigma factor RpoD-dependent consensus promoter sequence of phytoplasma. However, the genome-wide landscape of RNA transcripts, including non-coding RNAs (ncRNAs) and RpoD-independent promoter elements, was still unknown. In this study, we performed an improved RNA sequencing analysis for genome-wide identification of the transcription start sites (TSSs) and the consensus promoter sequences. We constructed cDNA libraries using a random adenine/thymine hexamer primer, in addition to a conventional random hexamer primer, for efficient sequencing of 5′-termini of AT-rich phytoplasma RNAs. We identified 231 TSSs, which were classified into four categories: mRNA TSSs, internal sense TSSs, antisense TSSs (asTSSs), and orphan TSSs (oTSSs). The presence of asTSSs and oTSSs indicated the genome-wide transcription of ncRNAs, which might act as regulatory ncRNAs in phytoplasmas. This is the first description of genome-wide phytoplasma ncRNAs. Using a de novo motif discovery program, we identified two consensus motif sequences located upstream of the TSSs. While one was almost identical to the RpoD-dependent consensus promoter sequence, the other was an unidentified novel motif, which might be recognized by another transcription initiation factor. These findings are valuable for understanding the regulatory mechanism of phytoplasma gene expression.
Introduction
T
Phytoplasmas (class Mollicutes, genus “Candidatus [Ca.] Phytoplasma”) are the bacterial plant pathogens that cause yield losses of various crops (The IRPCM Phytoplasma/Spiroplasma Working Team–Phytoplasma taxonomy group, 2004; Oshima et al., 2013; Maejima et al., 2014). Phytoplasmas are the obligate intracellular parasites that reside in the phloem tissues of infected plants and are transmitted by insect vectors in a persistent manner. Phytoplasma genomes are small (600–1,000 kb) and AT-rich (21–28%), and contain a limited number of genes (500–1,100 genes) (Oshima et al., 2004; Bai et al., 2006; Kube et al., 2008; Tran-Nguyen et al., 2008; Andersen et al., 2013). Phytoplasmas possess slightly larger genomes than mycoplasmas, which are closely related bacteria, due to repeated gene sequences called potential mobile units (PMUs) (Bai et al., 2006; Arashida et al., 2008). While phytoplasmas have lost more metabolic pathway genes than mycoplasmas, they possess multiple copies of transporter-related genes in PMUs for the absorption of nutrients from their host cells and adaptation to two different host cell environments (Oshima et al., 2004, 2013). We previously revealed that at least one-third of the genes of “Ca. P. asteris” onion yellows strain (OY-M) are differentially expressed in plants and insects (Oshima et al., 2011). However, the regulatory mechanisms of gene expression in phytoplasmas are poorly understood.
Complete genome sequence analyses of phytoplasmas revealed the presence of two types of sigma factors, RpoD and FliA (Oshima et al., 2004; Bai et al., 2006; Kube et al., 2008; Tran-Nguyen et al., 2008; Andersen et al., 2013). We previously reported that RpoD of OY-M regulates several housekeeping, virulence, and host–phytoplasma interaction genes of OY-M, using an in vitro transcription assay, and determined the consensus promoter sequence for RpoD (Miura et al., 2015). Although hundreds of candidate promoter sequences regulated by RpoD were predicted in the OY-M genome, their promoter activities in vivo remain to be elucidated. Moreover, the presence of RpoD-independent promoter elements is unknown. In this study, we applied RNA-Seq technology for genome-wide identification of TSSs and promoter elements of OY-M phytoplasma.
Materials and Methods
Plant materials
The “Ca. P. asteris” OY strain was isolated in Saga Prefecture, Japan, in May 1982 (Shiomi et al., 1998). A derivative line of OY (OY-M) was maintained in garland chrysanthemum (Glebionis coronaria), using the leafhopper vector Macrosteles striifrons (Oshima et al., 2001). The OY-M-infected plants were maintained at 25°C in a greenhouse, with a 16-h light/8-h dark photoperiod.
RNA extraction and sequencing
Total RNA was extracted from OY-M-infected plants using ISOGEN (Nippon Gene), and treated with RNase-free recombinant DNase I (TaKaRa). To digest rRNAs of phytoplasmas and plants, total RNA was treated with terminator 5′-phosphate-dependent exonuclease (TEX; Epicentre). The mRNA-enriched total RNA was treated with RNA 5′ polyphosphatase (Epicentre) to remove pyrophosphates from the 5′ ends of bacterial mRNA for adapter ligation.
The cDNA library was constructed using the TruSeq Small RNA Sample Prep Kit (Illumina) as follows. The 5′ RNA adapter was ligated to the 5′ phosphate of the pretreated total RNA. First-strand cDNA synthesis was then performed with an adapter-tagged random hexamer primer (N6 primer, 5′-
Mapping of RNA-Seq reads
The RNA adapter sequence and reads <20 bp were trimmed by the program cutadapt ver. 1.8.3 (Martin, 2011). Trimmed reads were mapped to the reference genome sequence of OY-M (accession number AP006628), using the program Bowtie2 version 2.2.6 (Langmead and Salzberg, 2012). The number of reads mapped to rRNAs or tRNAs were counted using the program HTSeq ver. 0.6.1p2 (Anders et al., 2015). The RNA-Seq reads were deposited in the DNA Data Bank of Japan Sequence Read Archive (
TSS annotation and classification
We annotated putative TSS positions where at least three reads share common 5′ ends. The TSSs were classified into the four categories as follows: mRNA TSSs (mTSSs)—TSSs located within 500 bp upstream of ORFs; internal TSSs (iTSSs)—TSSs located in ORFs in the sense orientation; antisense TSSs (asTSSs)—TSSs located in ORFs in the antisense orientation; and orphan TSSs (oTSSs)—all TSSs that did not fall in the above categories. The positions of the four kinds of TSSs in the OY-M were visualized using DNAPlotter (Carver et al., 2009). Operons in the OY-M genome were predicted using the Prokaryotic Operon DataBase (Taboada et al., 2012) (available at
Identification of de novo promoter motifs
To identify de novo promoter motifs, sequences representing 21 bp upstream of all TSSs, including the TSS nucleotides, were used as input for the program MEME ver. 4.11.1 (Bailey et al., 2006), with the following parameters: -dna -maxsize 100000 -mod zoops -nmotifs 10 -minsites 5 -maxsites 231 -minw 6 -maxw 21. Motifs with an E-value of <1 were examined further. Additionally, we searched 6 to 21-base long motifs serially by changing the “-maxw” parameter, to avoid missing TSSs with motifs. We started to search motifs with six-base long, since the length of the promoter sequences in bacteria is usually at least six-base long (Wösten, 1998). Since the genomes of phytoplasmas have repeated sequences (Bai et al., 2006; Arashida et al., 2008), motifs related to these repeated sequences were omitted. Sequence logos (Schneider and Stephens, 1990) were illustrated with WebLogo (Crooks et al., 2004) (available at
Comparative analysis
For comparative analysis with other phytoplasmas and closely related bacteria, the following genome sequences were used in this study; “Ca. P. asteris” AYWB strain (GenBank accession number CP000061), “Ca. P. australiense” rp-A (GenBank accession number AM422018) and NZSb11 (GenBank accession number CP002548), “Ca. P. mali” AT strain (GenBank accession number CU469464), “Ca. P. solani” 231/09 (GenBank accession number FO393428) and 284/09 strain (GenBank accession number FO393427), Acholeplasma laidlawii PG-8A strain (GenBank accession number CP000896), and Bacillus subtilis subsp. subtilis 168 strain (GenBank accession number AL009126).
Results
Genome-wide mapping and classification of TSSs
For genome-wide identification of TSSs of OY-M phytoplasma, we performed an RNA-Seq analysis, following the protocol used in previous studies (Sharma et al., 2010; Filiatrault et al., 2011; Schlüter et al., 2013; Sass et al., 2015; Čuklina et al., 2016). Total RNA extracted from garland chrysanthemum (G. coronaria) infected by OY-M phytoplasma was used for the construction of cDNA libraries. Total RNA was treated with TEX for the digestion of 5′ monophosphorylated RNAs (e.g., rRNAs, tRNAs, and degraded RNAs). For the selective construction of cDNA libraries of phytoplasma RNAs, the sample was then treated with 5′ polyphosphatase, which removes phosphates from 5′ triphosphorylated RNAs, such as mRNAs of bacteria, but not from 5′-capped RNAs, such as mRNAs of plants. After the ligation of RNA adapters to the 5′ ends of dephosphorylated RNAs, cDNA syntheses were performed to amplify cDNA libraries, using two random primers: the conventional adapter-tagged random hexamer primer (N6 primer) or the adapter-tagged random adenine/thymine hexamer primer (W6 primer). The W6 primer was used for efficient cDNA synthesis of phytoplasma RNAs, as phytoplasmas have AT-rich genomes. The construction of each cDNA library, using the N6 (N6 primer library) and W6 (W6 primer library) primers, was performed twice for technical replication. The libraries were sequenced on the MiSeq (Illumina).
After trimming adapter sequences, we obtained 3,052,036 and 5,299,788 reads, ranging from 20 to 50 bp, from the N6 and W6 primer libraries, respectively (Table 1). Overall, 4.52% and 11.67% of the total reads from N6 and W6 primer libraries, respectively, were mapped to the genome of the OY-M phytoplasma (Table 1). Of the reads mapped to the OY-M genome, 96.5% (132,912 reads) and 96.2% (595,143 reads) from the N6 and W6 primer libraries, respectively, were mapped to rRNAs or tRNAs. In other words, 3.5% (4,892 reads) and 3.8% (23,467 reads) were mapped to the OY-M genome excepting rRNAs and tRNAs.
The construction of each RNA-Seq library was performed twice for technical replication.
Number of RNA-Seq reads mapped to the OY-M genome/number of total RNA-Seq reads. Numbers in parentheses indicate percentages of RNA-Seq reads mapped to the OY-M genome.
OY-M, onion yellows strain; RNA-Seq, RNA sequencing.
In the previous studies of culturable bacteria, TEX-untreated library was used as a control (Sharma et al., 2010; Filiatrault et al., 2011; Schlüter et al., 2013; Sass et al., 2015; Čuklina et al., 2016). Therefore, we also constructed the TEX-untreated library using the W6 primer and the library was sequenced (Supplementary Table S1; Supplementary Data are available online at
We annotated the TSS positions, where at least three reads share common 5′ ends. Overall, 231 loci were annotated as the TSSs, two of which (amp and pam486) were previously annotated (Miura et al., 2015), whereas the other 229 were novel. Of the 231 loci, 147 (63.6%) were observed in at least two technical replications of the N6 or W6 libraries.
Since the classification of TSSs differed in each previous study (Sharma et al., 2010; Filiatrault et al., 2011; Sass et al., 2015; Čuklina et al., 2016), we simply classified the TSSs into four categories (Fig. 1A and Supplementary Fig. S1): mTSSs located within 500 bp upstream of ORFs in the sense orientation, iTSSs located in ORFs in the sense orientation, asTSSs located in ORFs in the antisense orientation, and the other TSSs classified as oTSSs. Each category of TSSs was further analyzed as follows.
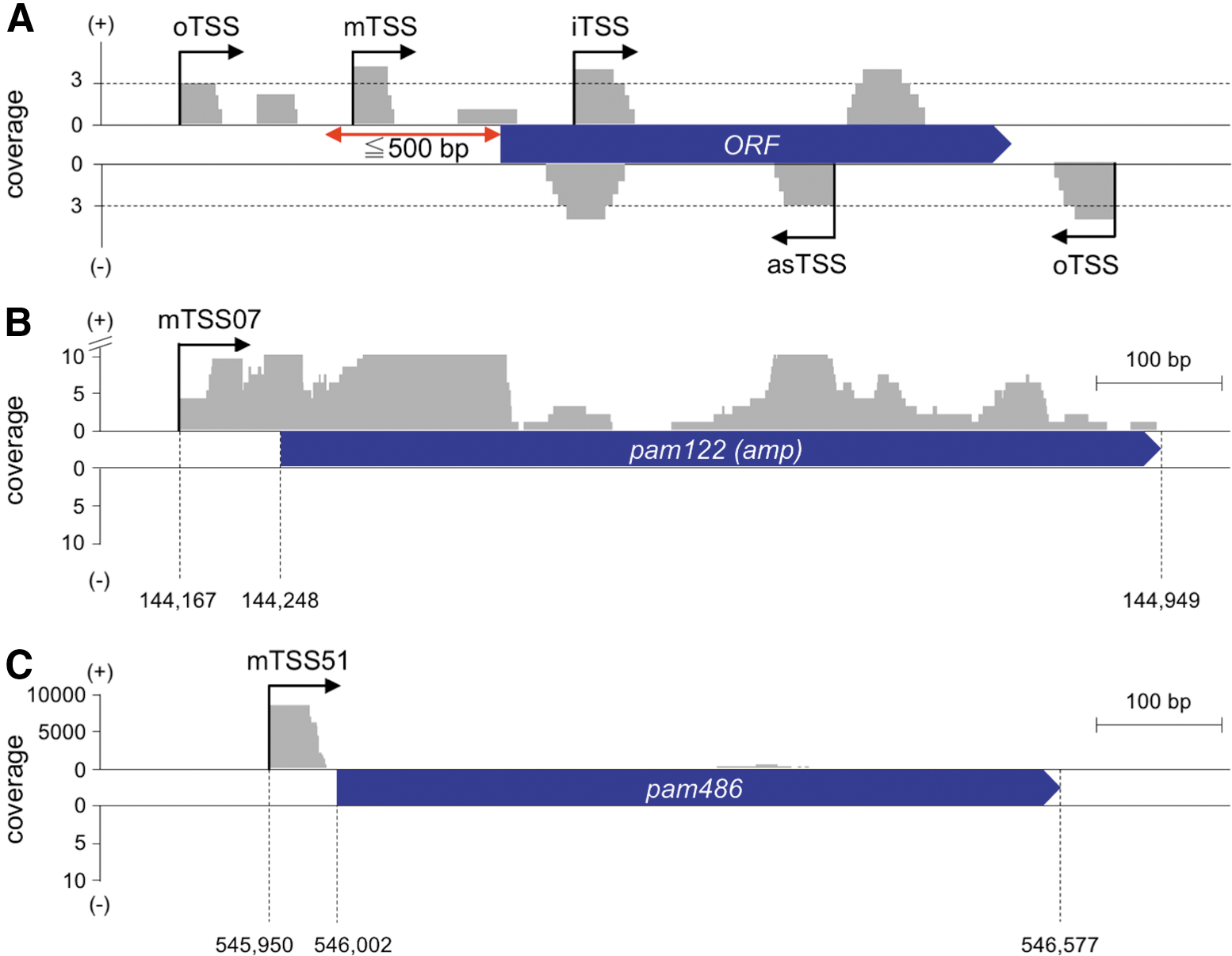
Mapping of RNA-Seq reads to the OY-M genome and categorization of TSSs. The loci where at least three reads share common 5′ ends were annotated as TSSs.
mRNA TSSs
Eighty-two TSSs were classified as mTSSs (Supplementary Table S2). The RNA transcripts from mTSSs were expected to serve as mRNAs of downstream ORFs. Two of the 82 mTSSs, located upstream of amp or pam486 (mTSS07 and mTSS51, shown in Fig. 1B, C, respectively), were at the same positions, as determined by 5′ rapid amplification of cDNA end (5′ RACE) analyses in our previous study (Miura et al., 2015). Thirty-four mTSSs were located upstream of putative operons (Supplementary Table S2) predicted by ProOpDB (Taboada et al., 2012). Of the previously annotated 791 ORFs of OY-M phytoplasma (Oshima et al., 2004), including downstream ORFs in these 34 operons, 131 ORFs (16.6%) were located downstream of 82 mTSSs. The average distance from the mTSS to the translation start codon was 187 bp, and 52 of 82 mTSSs (63.4%) were located within 200 bp upstream of the translation start codon (Table 2). Eight ORFs (pam160, pam207, pam264, pam267, pam312, pam486, pam530, pam602) possessed two mTSSs located upstream of each mTSS (Supplementary Table S1).
The previous study analyzed only TSSs located within 250 bp of the translation start codon.
n.t., not tested; mTSS, mRNA TSS; TSSs, transcription start sites.
Internal TSSs
Eighty-eight TSSs were classified as iTSSs (Supplementary Table S3). The iTSS of pam135 (iTSS07) was located just on the annotated GTG (valine) start codon. This result indicates that the RNA transcript from iTSS07 lacks a 5′ untranslated region, where ribosomal subunits could bind. However, we found that pam135 possesses an ATG (methionine) start codon 9 bp downstream of GTG (Fig. 2A). Recently, the same annotation of pam135 that starts from the ATG codon 9 bp downstream of the original one (accession number NC005303.2) was reported, using the National Center for Biotechnology Information Prokaryotic Genome Annotation Pipeline (NCBI PGAP) (Tatusova et al., 2016).

The iTSS located on the translation start codon of pam135.
Although the function of PAM135 is unknown, other phytoplasmas and closely related bacteria also possess the homologous gene of pam135. The homologous gene of pam135 in Staphylococcus aureus has been reported to be involved in the recognition of cyclic-di-AMP (Müller et al., 2015), which is a bacterial secondary messenger of signal transduction (Kolb et al., 1993). Therefore, we next analyzed conservation of the start codon of the pam135 homologs in phytoplasmas and closely related bacteria. The downstream ATG codon was also found in the pam135 homologs of other phytoplasmas and closely related bacteria (Fig. 2B). The amino acids in each position of PAM135 homologs were more conserved downstream of the conserved ATG codon (Fig. 2B). These data suggest that PAM135 and its homologs are likely to be translated from the conserved ATG codon. Although other genes with iTSSs were not reannotated by the NCBI PGAP, most iTSSs, including that of pam135 (86 of 88 TSSs), possessed at least one putative alternative start codon (ATG, TTG, or GTG) downstream. Therefore, these ORFs should be analyzed further to identify legitimate start codons.
Antisense TSSs
Thirty-one TSSs were classified as asTSSs (Supplementary Table S4). The RNA transcripts from asTSSs were considered as ncRNAs, as no ORFs were predicted downstream of asTSSs. An example of the asTSS (asTSS22) in pam667 (clpX) is shown in Figure 3A. In pam667 (total length of 1,131 bp), an antisense RNA would be transcribed from the position of 497 bp downstream of the start codon. The presence of asTSSs implies the genome-wide presence of antisense ncRNAs in OY-M phytoplasma. In phytoplasmas, we revealed the genome-wide presence of antisense ncRNAs for the first time.

Examples of asTSS and oTSS.
Orphan TSSs
Thirty TSSs were classified as oTSSs (Supplementary Table S5). Eleven oTSSs were located upstream of ORFs, in the same orientation, and 19 oTSSs were located downstream of ORFs, in a different orientation (Supplementary Table S5). From both data of the TEX-treated and the TEX-untreated library, we confirmed that there are mapped reads downstream of all oTSS, which would be derived from degraded transcripts from the oTSSs. This result supports the reliability of oTSSs annotated in this study. We found putative new ORFs located downstream of two oTSSs (oTSS14 and oTSS28), and these ORFs were also predicted by the NCBI PGAP (rs02680 and rs04245; Fig. 3B, C, respectively). Although the functions of these proteins are unknown, the homologous genes are encoded in the genome of the “Ca. P. asteris” AYWB strain (aywb_319 and aywb_063, respectively; Bai et al., 2006). Therefore, these oTSSs might, in fact, act as mTSSs. We also found four oTSSs to be located within putative new ORFs, reported by NCBI PGAP, in the antisense orientation (Table 3), implying that these four oTSSs might act as asTSSs. Therefore, the actual number of oTSSs might be 24. Since no ORFs were predicted to be located downstream from these 24 oTSSs, the RNA transcripts from the 24 oTSSs were considered to be ncRNAs. Considering the results for asTSSs and oTSSs, more than 25.5% (59 of 231 TSSs) could serve as TSSs for ncRNAs. This result indicates that several phytoplasma RNAs act as ncRNAs, which may regulate phytoplasma gene expression.
Defined in Supplementary Table S5.
Numbers indicate nucleotide positions relative to the OY-M genome.
+ and − indicate that the transcript from the oTSS is in the sense and antisense orientations of the OY-M genome, respectively.
Annotated by the NCBI PGAP (Tatusova et al., 2016).
NCBI PGAP, National Center for Biotechnology Information Prokaryotic Genome Annotation Pipeline; ORF, open reading frame; oTSS, orphan TSS.
Identification of promoter motifs
In the regulation of bacterial transcription, promoter sequences located upstream of TSSs are recognized by sigma factors. Previously, we identified a consensus promoter sequence recognized by RpoD of OY-M phytoplasma, based on in vitro transcription analysis (Miura et al., 2015). To identify other putative promoter sequences, we obtained the information on the upstream sequences of all of the TSSs from the OY-M genome sequence (accession number AP006628) and submitted them to MEME, a program for de novo motif discovery (Bailey et al., 2006). Since the consensus −10 promoter element of RpoD is more conserved than the consensus −35 promoter element (Miura et al., 2015), we used upstream sequences from −20 to +1 of TSSs to identify the consensus sequences other than the RpoD-dependent one. The MEME found two motifs located upstream of 71 TSSs (Fig. 4A, B and Tables 4 and 5). Motif 1 was found in 41 sites (Table 4), including upstream of amp and pam486, which are transcriptionally regulated by RpoD (Miura et al., 2015). Motif 1 was identified to be located upstream of 22 of 82 mTSSs, 7 of 88 iTSSs, 5 of 31 asTSSs, and 7 of 30 oTSSs in this study. In motif 1, we found the consensus −10 promoter element (5′-TAtAAT-3′) and an extended −10 motif (5′-TnTG-3′; Fig. 4A), which were almost identical to the RpoD-dependent consensus promoter sequence identified in our previous study (Miura et al., 2015). We also found the −35 promoter element and an A-rich region located upstream of these TSSs, which were less conserved than the consensus −10 promoter element and the extended −10 motif (Supplementary Fig. S2A), but were similar to those of the RpoD-dependent consensus promoter sequence (Miura et al., 2015). Notably, motif 1 was found in the upstream regions of the mTSSs of pam153 (mTSS10) and pam734 (norM, mTSS80), which had been predicted as an RpoD-dependent promoter in our previous study (Miura et al., 2015) by in silico tool, RSA-tools (van Helden, 2003) using the experimentally determined RpoD-dependent consensus promoter sequence. These results indicate that motif 1 would be recognized by RpoD. Motif 2 was found upstream of 30 TSSs (Fig. 4B and Table 5). Motif 2 was identified to be located upstream of 5 of 82 mTSSs, 7 of 88 iTSSs, 10 of 31 asTSSs, and 8 of 30 oTSSs. Motif 2 was not located upstream of RpoD-regulated genes (Miura et al., 2015). In the upstream regions of the TSSs with motif 2, regions around −10 were relatively conserved and T-rich (Supplementary Fig. S2B).

Conserved motifs located upstream of TSSs. Sequence logos of
Defined in Supplementary Tables S2–S5.
Numbers indicate nucleotide positions relative to the OY-M genome.
+ and − indicate that the transcript from the TSS is in the sense and antisense orientation of the OY-M genome, respectively.
ORFs corresponding to the mTSSs.
Annotated in our previous study (Oshima et al., 2004).
Confirmed to be regulated by RpoD using the in vitro assay in our previous study (Miura et al., 2015).
Predicted to be regulated by RpoD in our previous study (Miura et al., 2015).
Defined in Supplementary Tables S2–S5.
Numbers indicate nucleotide positions relative to the OY-M genome.
+ and − indicate that the transcript from the TSS is in the sense and antisense orientations of the OY-M genome, respectively.
ORFs corresponding to the mTSSs.
Annotated in our previous study (Oshima et al., 2004).
In addition, we analyzed whether the two motifs were conserved among phytoplasmas. Each motif was also found in the upstream region of homologous genes in other phytoplasma genomes (Fig. 5 and Supplementary Fig. S3). Therefore, these motifs could be universally involved in transcriptional regulation of phytoplasmas.
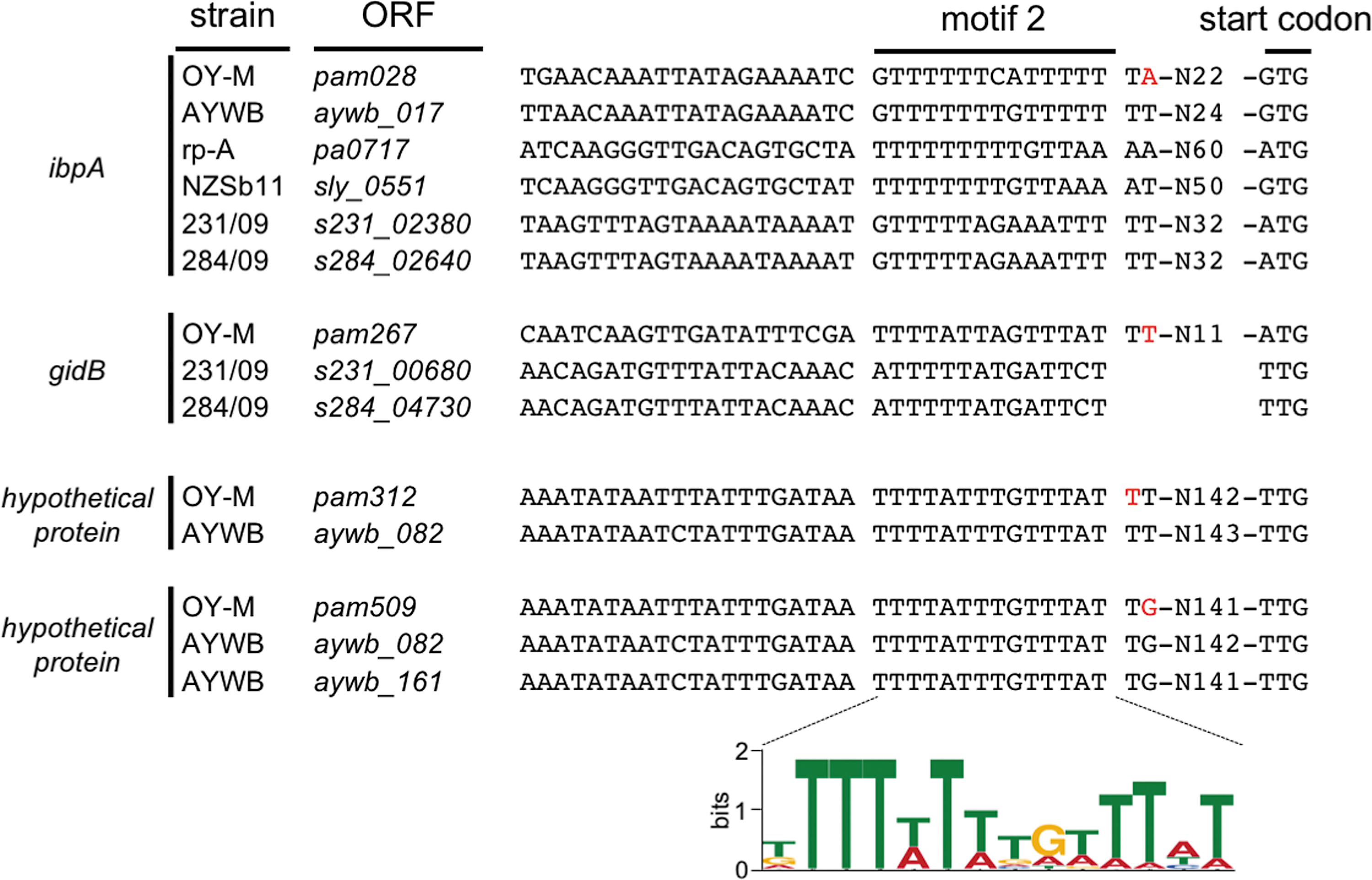
Conservation of motif 2 in phytoplasmas. Red letters indicate TSSs identified in this study. Numbers on the right side show the distance to the start codon. The sequence logo was illustrated with WebLogo. Y-axes indicate the sequence conservation. Color images available online at
Discussion
Recent advances in RNA-Seq technology have contributed to understanding bacterial gene transcription. However, although genome-wide TSSs and promoter consensus sequences have been reported in many culturable bacteria (Mendoza-Vargas et al., 2009; Sharma et al., 2010; Filiatrault et al., 2011; Schlüter et al., 2013; Sass et al., 2015; Čuklina et al., 2016), genome-wide analyses on TSSs and promoter consensus sequences have not been reported in unculturable bacteria, due to the inefficiency in excluding unspecific RNAs from hosts or environments. In the previous studies of the transcriptome analysis of phytoplasmas, only <0.1% of the reads were mapped to the phytoplasma genomes (Abbá et al., 2014; Siewert et al., 2014). Therefore, the regulatory mechanism of gene expression in these bacteria is poorly understood. In this study, we thus developed an improved genome-wide RNA-Seq analysis to identify TSSs and promoter consensus sequences of OY-M phytoplasma, an unculturable bacterium.
In the construction of the cDNA library for RNA-Seq, we used the W6 primer, as phytoplasmas have AT-rich genomes (Oshima et al., 2004; Bai et al., 2006; Kube et al., 2008; Tran-Nguyen et al., 2008; Andersen et al., 2013). The RNA-Seq reads from libraries constructed using the W6 primer were more frequently (at least twice) mapped to the phytoplasma genome than those using the N6 primer. Since cDNA synthesis is biased toward AT-rich sequences, our results suggest that the use of a random W6 primer would not be suitable for transcriptome expression analysis by RNA-Seq. However, the use of the modified random W6 primer is helpful for TSS analysis in phytoplasmas, and for analyses in other unculturable bacteria, such as the plant pathogen “Ca. Liberibacter asiaticus” (Duan et al., 2009) and the vertebrate pathogen “Ca. Mycoplasma haemolamae” (Guimaraes et al., 2012), as many obligate intracellular bacteria possess AT-rich genomes (Moran, 2002; Moran et al., 2008).
In addition to the use of W6 primer for the construction of the cDNA library, total RNA was treated with TEX for the digestion of 5′ monophosphorylated RNAs (e.g., rRNAs, tRNAs, and degraded RNAs). Without the TEX treatment, most of sequence reads mapped on ORFs would be derived from degraded mRNAs, whereas a part of them would be derived from 5′-ends of iTSSs or asTSSs. It was considered that the TEX treatment is useful for decreasing the degraded mRNAs, since it decreased the percentages of reads mapped on ORFs in the non-rRNA/tRNA-mapped reads from 53.3% to 35.0%. Moreover, the TEX treatment increased the percentage of intergenic-mapped reads in the non-rRNA/tRNA-mapped reads from 46.7% to 65.0%, enabling us to obtain sequence reads derived from mTSSs and oTSSs.
In this study, we annotated 231 loci as putative TSSs. Although the enrichment of 5′ ends of mRNAs using TEX and RNA 5′ polyphosphatase is not complete, as previously described (Schlüter et al., 2013), 63.6% of the annotated TSSs were observed in at least two replications of the N6 and/or W6 primer libraries. This result supports the validity of the TSS annotation. We categorized 231 TSSs into four groups: mTSSs, iTSSs, asTSSs, and oTSSs. In total, we found 82 mTSSs that could be related to the transcription of 131 ORFs, including downstream ORFs in putative operons of OY-M phytoplasma. On the other hand, of the previously annotated 791 ORFs (Oshima et al., 2004), we could not find mTSSs of the 660 other ORFs. The number of the previous annotated genes of OY-M includes 46 pseudogenes. Additionally, there are repeated gene sequences called PMUs in the phytoplasma genome (Bai et al., 2006; Arashida et al., 2008). Of the annotated 791 ORFs of OY-M phytoplasma, 147 ORFs (18.6%) were multiple redundant genes (Arashida et al., 2008). It is unclear whether all of the multiple redundant genes are actually expressed. It has been reported that although OY phytoplasma possess 11 copies of thymidylate kinase (tmk) genes, 1 of them does not possess the thymidylate kinase activity (Miyata et al., 2003). Therefore, the actual number of the genes of OY-M phytoplasma would be <791. Moreover, OY-M phytoplasma alters the gene accumulation level dramatically upon host switching between plants and insects (Oshima et al., 2011), which may be the other reason why mTSSs of the 660 other ORFs were not detected in this study.
The average distance from the mTSS to the translation start codon was 187 bp, and more than 60% of the mTSSs were located within 200 bp of the translation start codon. These findings were similar to those in a study of Mycoplasma hyopneumoniae, which is closely related to phytoplasmas (Weber et al., 2012). We previously reported that the consensus promoter sequences recognized by RpoD of phytoplasmas are also similar to those of M. hyopneumoniae (Miura et al., 2015). Thus, the regulatory mechanism of transcription initiation might be common in the Mollicutes class, to which phytoplasmas and mycoplasmas belong. In addition to the example of rrnB described in our previous report (Miura et al., 2015), we found that several ORFs possessed two mTSSs located upstream of each mTSS in this study. The multiple mTSSs located upstream of the same ORFs are also reported in other bacteria (Schlüter et al., 2013; Sass et al., 2015), and are thought to contribute to the increase of transcription (Young and Steitz, 1979). Therefore, phytoplasmas might also use the multiple mTSSs for the regulation of gene expression levels.
Previous studies reported the usefulness of iTSSs for improvement of the annotation of ORFs (Sharma et al., 2010; Schlüter et al., 2013). In this study, we discovered that pam135 possessed the iTSS just on the annotated start codon and the alternative start codon located downstream of the iTSS. We also found that most of the iTSSs possessed alternative downstream start codons. There is also a possibility that pam135 possesses a leaderless mRNA, as reported in other bacteria (Sharma et al., 2010; Schlüter et al., 2013). While the validity and function of each iTSS should be individually checked in future studies, the information on iTSSs would be useful in confirmation of the genome annotation of unculturable bacteria.
Bacterial ncRNAs have roles in the regulation of gene expression (Waters and Storz, 2009; Storz et al., 2011). A previous study reported one ncRNA in Flavescence dorée phytoplasma (Abbá et al., 2014), but genome-wide insights into the ncRNAs of phytoplasmas were lacking. In addition, since the reported phytoplasma ncRNA was related to the catalytically active RNA of a group II intron, which is a genetic element involved in self-splicing and mobility (Dai and Zimmerly, 2002; Wei et al., 2008), ncRNAs that would be involved in the regulation of gene expression have not been found in phytoplasmas.
In this study, we identified the genome-wide presence of asTSSs and oTSSs in the OY-M phytoplasma. Several oTSSs were located nearby, downstream of ORFs, in the antisense orientation. It will be necessary to assess whether oTSSs could act as mTSSs of ORFs located more than 500 bp downstream since there are mapped reads downstream of oTSSs, which would be derived from degraded RNAs. However, several RNAs transcribed from these oTSSs might act in a manner similar to those from asTSSs, as cis-regulatory elements. These ncRNAs might also act as trans-regulatory elements, since the genomes of phytoplasmas have repeated gene sequences, called PMUs (Bai et al., 2006; Arashida et al., 2008). Thus, phytoplasmas also seem to utilize ncRNAs for the regulation of gene expression, like other bacteria. Further analyses, including determination of the complete sequences of ncRNAs by 3′ RACE, are necessary to identify the roles of these ncRNAs.
The upstream sequences of TSSs are usually considered as promoter sequences recognized by sigma factors. In this study, we conducted a genome-wide analysis of de novo consensus motifs and found two consensus motif sequences (motifs 1 and 2). Although 71 of the 231 TSSs possessed consensus motif sequences located upstream of TSSs, we could not find consensus motif sequences located upstream of the other 160 TSSs. This might be because of the E-value threshold to avoid false positives. Therefore, some of the 160 TSSs would have motif 1 or 2 in their upstream regions.
Motif 1 possessed the consensus −10 element (5′-TAtAAT-3′) and an extended −10 motif (5′-TnTG-3′). Since motif 1 was found upstream of amp and pam486, which were transcriptionally regulated by RpoD, and possessed the RpoD-consensus promoter elements (Miura et al., 2015), it is most likely to be the consensus promoter sequences recognized by RpoD. Among hundreds of candidate promoter sequences recognized by RpoD in the OY-M genome predicted based on in silico tools (Miura et al., 2015), we detected only four promoter sequences (amp, pam486, pam153, and norM) in this study. This might be because of the low-level accumulation of the other RNAs. On the other hand, we newly found consensus promoter sequences that seem to be recognized by RpoD at 37 sites. In comparison with the −10 promoter element, the −35 promoter element was less conserved. This result is consistent with our previous study, which also mentioned that the gap lengths between the two elements varied 17–19 bp (Miura et al., 2015). The other previous study also reported the less conservation of the −35 promoter element in mycoplasmas, closely related to phytoplasmas (Weber et al., 2012). We also found motif 1 upstream of homologous genes in phytoplasma genomes other than OY-M. Therefore, RpoD should regulate the homologous genes similarly in phytoplasmas.
Motif 2, the T-rich motif in the −10 region, was not similar to the RpoD-dependent promoter motif, nor to any promoter consensus sequences previously reported in other bacteria (Gruber and Gross, 2003). Although the AT-richness of phytoplasma genomes could happen to provide a T-rich motif like motif 2, we also found motif 2 in the upstream regions of homologous genes in other phytoplasmas. Although some strains possess motif 2 in different loci upstream from start codons, a previous study reported that the distance from the promoter sequence to the start codon could differ in the homologous genes of closely related bacteria (Song et al., 2007). These results support the existence of this motif in phytoplasmas. Therefore, motif 2 would be recognized by transcription factors other than RpoD. Since phytoplasmas have only two sigma factors, RpoD and FliA (Oshima et al., 2004; Bai et al., 2006; Kube et al., 2008; Tran-Nguyen et al., 2008; Andersen et al., 2013), motif 2 might be recognized by FliA. In the upstream regions of TSSs with motif 2, the −35 region was less conserved than the −10 region. This would be consistent with the lack of domain 4 in the phytoplasma FliA, for recognition of the −35 promoter elements (Miura et al., 2015). Since the phytoplasmal genes directly regulated by FliA are not clear yet, the in vitro transcription assay would be helpful to reveal, in detail, the role of motif 2 and its relationship to the alternative sigma factor FliA.
Our data suggest the presence of mRNAs and ncRNAs, and their regulation by sigma factors in phytoplasmas. Phytoplasma possesses multiple-copy genes in PMUs, such as transporter-related genes (Oshima et al., 2004, 2013), and alter their expression in response to host switching (Oshima et al., 2011). These genes would be regulated intricately by two sigma factors and many ncRNAs. Further analyses on the function of FliA and ncRNAs would reveal the mechanism of phytoplasma gene regulation in host switching between plants and insects.
Conclusion
In this study, for the first time, we performed genome-wide analysis on TSSs of phytoplasmas. Using an improved RNA-Seq technology, we identified 231 TSSs. The average distance from the translation start codon to the mTSS was similar to that of M. hyopneumoniae. The information on iTSSs was helpful for appropriate annotation of ORFs. The presence of asTSSs and oTSSs indicated that the OY-M phytoplasma has ncRNAs. Using the sequences upstream of TSSs, we identified a consensus motif regulated by RpoD, and a novel consensus motif that might be recognized by another transcription factor. Our data of TSSs and consensus motifs of OY-M phytoplasma should be helpful for understanding the regulation of gene expression in phytoplasmas.
Footnotes
Acknowledgment
This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (category “S” of Scientific Research Grant 25221201).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.