
Abstract
RNA-sequencing, a powerful tool, yields a comprehensive view of whole transcriptome. Intracerebral hemorrhage (ICH) is a devastating form of stroke. To date, RNA-sequencing analysis of ICH has not been reported. Peripheral blood mononuclear cells (PBMCs) were used as a source of mRNA for gene expression profile analysis in stroke. In this study, we performed transcriptome analyses for PBMCs from four ICH patients and four healthy volunteers on Illumina platform. We identified 4040 significantly differentially expressed genes (DEGs). Functional annotation of DEGs with DAVID Bioinformatics Resources indicated that genes associated with cell apoptosis, autophagy, cell–cell adhesion, inflammatory response, protein binding, positive regulation of gene expression, and signal transduction were most significantly enriched by DEGs. Gene set enrichment analysis identified 40 significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, including chemokine signaling, cytokine–cytokine receptor interaction, oxidative phosphorylation, and glutathione metabolism processes. These data point to a complex mechanism for ICH pathogenesis. Overall, the present study demonstrated an altered gene expression profile of PBMCs in response to acute ICH. Our study provided important information for understanding the molecular mechanisms of ICH pathogenesis at system-wide levels.
Introduction
S
Different mechanisms have been proposed for the pathogenesis of stroke, and there is growing evidence that inflammation accounts for the progression and as potential therapeutic targets for this disease (del Zoppo and Hallenbeck, 2000; Gong et al., 2000; Zhang et al., 2006; Wang et al., 2007). Inflammatory cells may contribute to the recovery from cerebral ischemia (Tang et al., 2001). Because brain tissue is rarely available for study, previous studies have used peripheral blood mononuclear cells (PBMCs) as a source of mRNA to examine the changes of gene expression following stroke by microarray analysis (Tang et al., 2001; Moore et al., 2005). With the advance of the next-generation sequencing technologies, RNA-sequencing has become a useful tool with higher sensitivity than microarray to analyze gene expression profile. A recent study has shown that differential alternative splicing differs between ICH, ischemic stroke, and control subjects by RNA-sequencing (Dykstra-Aiello et al., 2015). In the current study, we utilized next-generation RNA-sequencing to sequence the whole transcriptomes of PBMCs from ICH patients or healthy volunteers. The differentially expressed genes (DEGs) and biological pathway alteration were identified, which provided comprehensive information on the molecular mechanisms of ICH.
Materials and Methods
Study participants
Thirty ICH patients admitted to Xiangyang No.1 Hospital affiliated to Hubei University were enrolled in this study (Table 1). Deep hemorrhage (basal ganglia, deep white matter, or thalamus) was confirmed in the ICH patients by CT and/or magnetic resonance scans. The ICH patients enrolled had hypertension, but no vascular malformation, tumor, or aneurysm. We excluded patients with recent infection (within 3 months) or taking immunosuppressive or anti-inflammatory drugs; combined with major cardiac, hepatic, renal, autoimmune, or cancerous disease; or with a history of previous stroke. Thirty healthy volunteers who had no history of previous stroke or cardiovascular events were selected (Table 1). Peripheral blood samples were collected from healthy volunteers or patients within 3 h following ICH and 6 months after ICH. This study received ethical approval from the Ethics Committee of Xiangyang No.1 Hospital affiliated to Hubei University. All participants were given written informed consent before enrollment.
ICH, intracerebral hemorrhage.
Total RNA isolation and sequencing
The blood of ICH patients and healthy volunteers was drawn through the antecubital vein. PBMCs were separated by centrifugation at 300 g for 30 min on mononuclear/polynuclear cell-resolving medium (Flow Laboratories, Rockville, MD). Total RNA was then extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. DNA was removed by digestion with RNase-free DNase (Sigma, St. Louis, MO). RNA quality was evaluated by gel electrophoresis and ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE). cDNA sequencing libraries were prepared using Illumina's TruSeq Sample Preparation Kit (San Diego, CA). Single-end raw sequence reads (36-mer) were generated using an Illumina HiSeq3000 platform according to the standard protocol. All reads were aligned to human transcript reference sequences from the Ensembl and RefSeq database. The DEGs (more than 1.5-fold differentially expressed with p-value less than 0.05) were identified by DESeq package (Anders and Huber, 2010). The statistical analysis was carried out using R language (
Functional annotation and gene enrichment analysis
Functional analysis of the DEGS was performed by DAVID Bioinformatics Resources (Huang da et al., 2009) and gene set enrichment analysis (GSEA) (Subramanian et al., 2007) as previously described. gene ontology (GO) terms associated with DEGs were calculated using DAVID (
Real-time polymerase chain reaction
Total RNA was reverse transcribed with RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Rockford, IL). Real-time polymerase chain reaction (PCR) was carried out using SYBR-green PCR Master Mix (Thermo Scientific) on an ABI 7300 instrument (Applied Biosystems, Foster City, CA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as the internal control. All real-time PCR primers were listed in as follows:
SLC9A8 (Solute Carrier Family 9, Subfamily A, Member 8), 5′-AGAGGAGCACCATCTACAG-3′ and 5′-GGAGCACAGGAAGTATCAC-3′;
ADM (adrenomedullin), 5′-TTGCCACTTCGGGCTTCTC-3′ and 5′-ACATCAGGGCGACGGAAAC-3′;
IL10 (Interleukin 10), 5′-TGCTGGAGGACTTTAAGG-3′ and 5′-CTTGATGTCTGGGTCTTG-3′;
TNFRSF12A (Tumor Necrosis Factor Receptor Superfamily, Member 12A), 5′-CTCGCCCACTCATCATTC-3′ and 5′-GAGGCTCCCTTTCTGTTC-3′;
CCR1 (Chemokine [C-C Motif] Receptor 1), 5′-GGGTGAGATTCTGTGTTG-3′ and 5′-TGGGTAGTGGAAGTGTAG-3′;
MMP9 (matrix metalloproteinase 9), 5′-AAGGGCGTCGTGGTTCCAACTC-3′ and 5′-AGCATTGCCGTCCTGGGTGTAG-3′;
GPX1 (Glutathione peroxidase 1), 5′-AGTCGGTGTATGCCTTCTC-3′ and 5′-CTTCGTTCTTGGCGTTCTC-3′;
GGT5 (Gamma-Glutamyltransferase 5), 5′-AAGTTGAGGCGTGAGTTTGG-3′ and 5′-CAGATGGACAGATGGGTGAATG-3′;
CDK6 (Cyclin-Dependent Kinase 6), 5′-CTTCATTCACACCGAGTAG-3′ and 5′- ACGACCACTGAGGTTAGAG-3′;
CCND2 (Cyclin D2), 5′-TACTCTTCGCTTCTGGTATCTG-3′ and 5′-GGAATGTTGTGATGGGAATGTC-3′;
GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-AATCCCATCACCATCTTC 3′ and 5′-AGGCTGTTGTCATACTTC-3′.
Results
RNA-sequencing analysis
PBMCs were obtained from four patients within 3 h following ICH, while PBMCs from four healthy volunteers were used as control. RNA-sequencing was performed to study the transcriptomes of patients and healthy volunteers using the Illumina platform (Fig. 1). A total of 4040 genes, at least 1.5-fold differentially expressed with p-value less than 0.05, were identified. These DEGs included 2177 upregulated genes and 1863 downregulated genes in the transcriptomes of patients as compared with that of healthy volunteers (Supplementary Tables S1 and S2; Supplementary Data are available online at
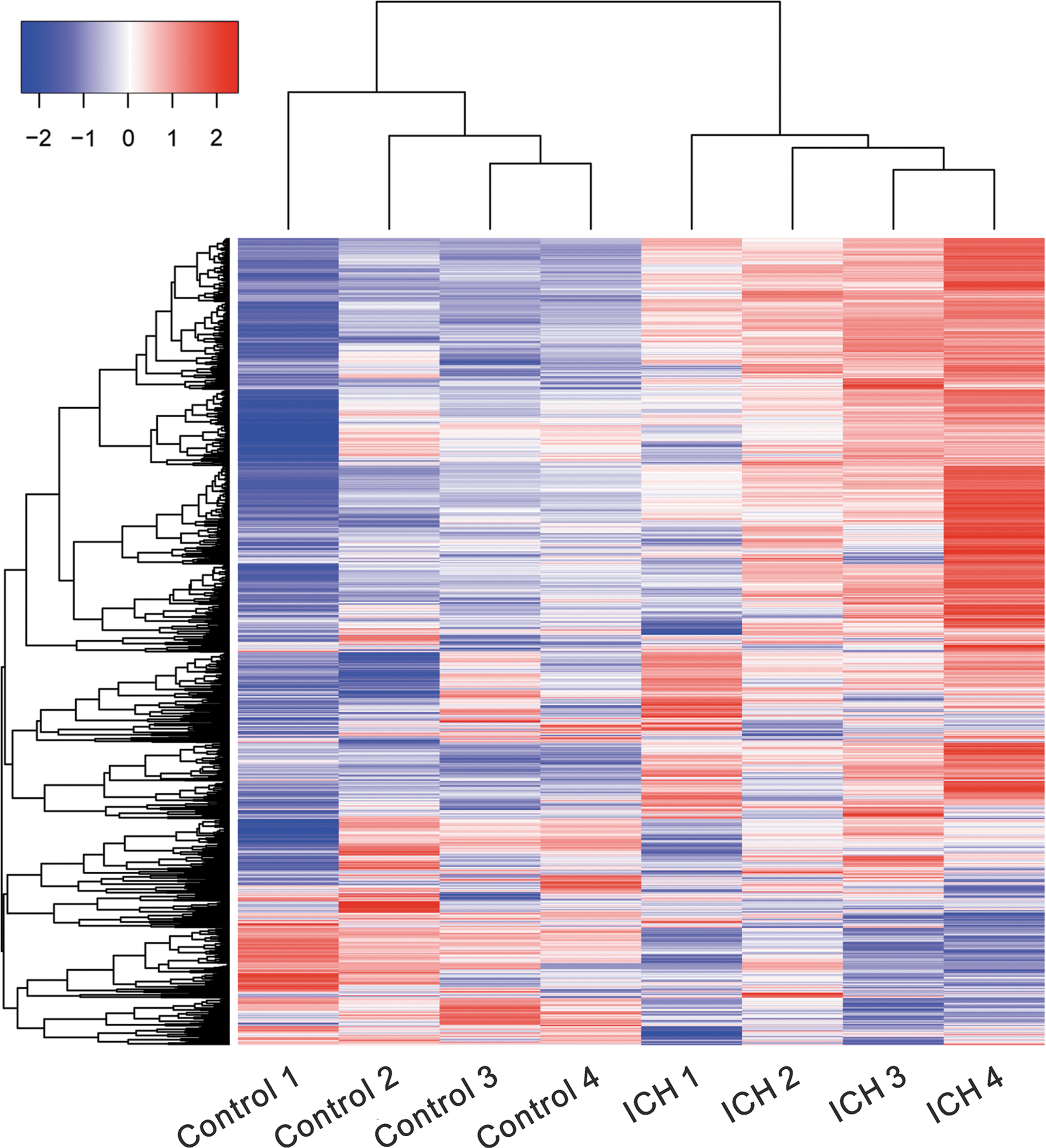
Heat map of genes expressed in PBMCs from four ICH patients and four controls. The color key represents RPKM normalized log2 transformed counts. Red indicates high expression, white indicates intermediate expression, and blue indicates low expression. ICH, intracerebral hemorrhage; PBMCs, peripheral blood mononuclear cells; RPKM, reads per kilobase per million mapped reads.
Functional annotation analysis of DEGs
To understand the biological significance of DEGs, we conducted a functional annotation of DEGs with DAVID Bioinformatics Resources and GSEA analysis. We found that 61 Go terms (upregulated, 49 terms; downregulated, 12 terms) were considered significant at a cutoff p-value <0.05 and false discovery rate <0.05 (Supplementary Table S3). The upregulated genes were mainly enriched in the terms of cell apoptosis, autophagy, cell–cell adhesion, inflammatory response, protein binding, positive regulation of gene expression, and signal transduction (GTPase and vascular endothelial growth factor receptor signaling). The downregulated genes were mainly enriched in the gene transcription and ATP binding. Through GSEA analysis, we identified 40 significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Supplementary Table S4 and Fig. 2) at a cutoff p-value <0.05, such as chemokine signaling (e.g., CCL7 [Chemokine (C-C Motif)] Ligand 7 and CCR1), cytokine–cytokine receptor interaction (e.g., TNFRSF12A, IL10, and CCL7), oxidative phosphorylation, and glutathione metabolism processes (e.g., GPX1 and GGT5).

Gene set enrichment analysis (GSEA) of signaling pathways strongly associated with ICH. Normalized enrichment score (NES) and p-value are shown as indicated. The heatmap compared gene expression between ICH and Control subjects. Gene expression is normalized for each row. Lower levels of expression were represented in shades of blue and higher expression in red. Figure text can be enlarged for ease of readability online at
Confirmation of expression measurements with real-time PCR
Then, to investigate whether the DEGs involved in the pathways were associated with the pathogenesis of ICH, genes in chemokine signaling (CCR1), cytokine–cytokine receptor interaction (IL10 and TNFRSF12A), glutathione metabolism processes (GPX1 and GGT5), ion exchange (SLC9A8), and cell proliferation (CDK6 and CCND2) were selected to further validate. Moreover, two genes, which have been identified as DEGs in previous studies on stroke [ADM (Moore et al., 2005) and MMP9 (Power et al., 2003)] were also selected.
PMBCs were obtained form 30 healthy controls, and 30 ICH patients at 3 h following ICH and at 6 months after ICH (Recovery). Real-time PCR was performed to detect the mRNA expression of the selected eight upregulated genes and two downregulated genes. As shown in Figure 3, within 3 h following ICH, the expression changes of detected genes were consistent between the results of RNA-sequencing and real-time PCR. Moreover, the upregulated genes were significantly decreased at 6 months after ICH, while the downregulated genes were notably increased at 6 months after ICH in most patients (Fig. 4). These results indicated the importance of the detected genes in the pathogenesis and recovery of ICH.
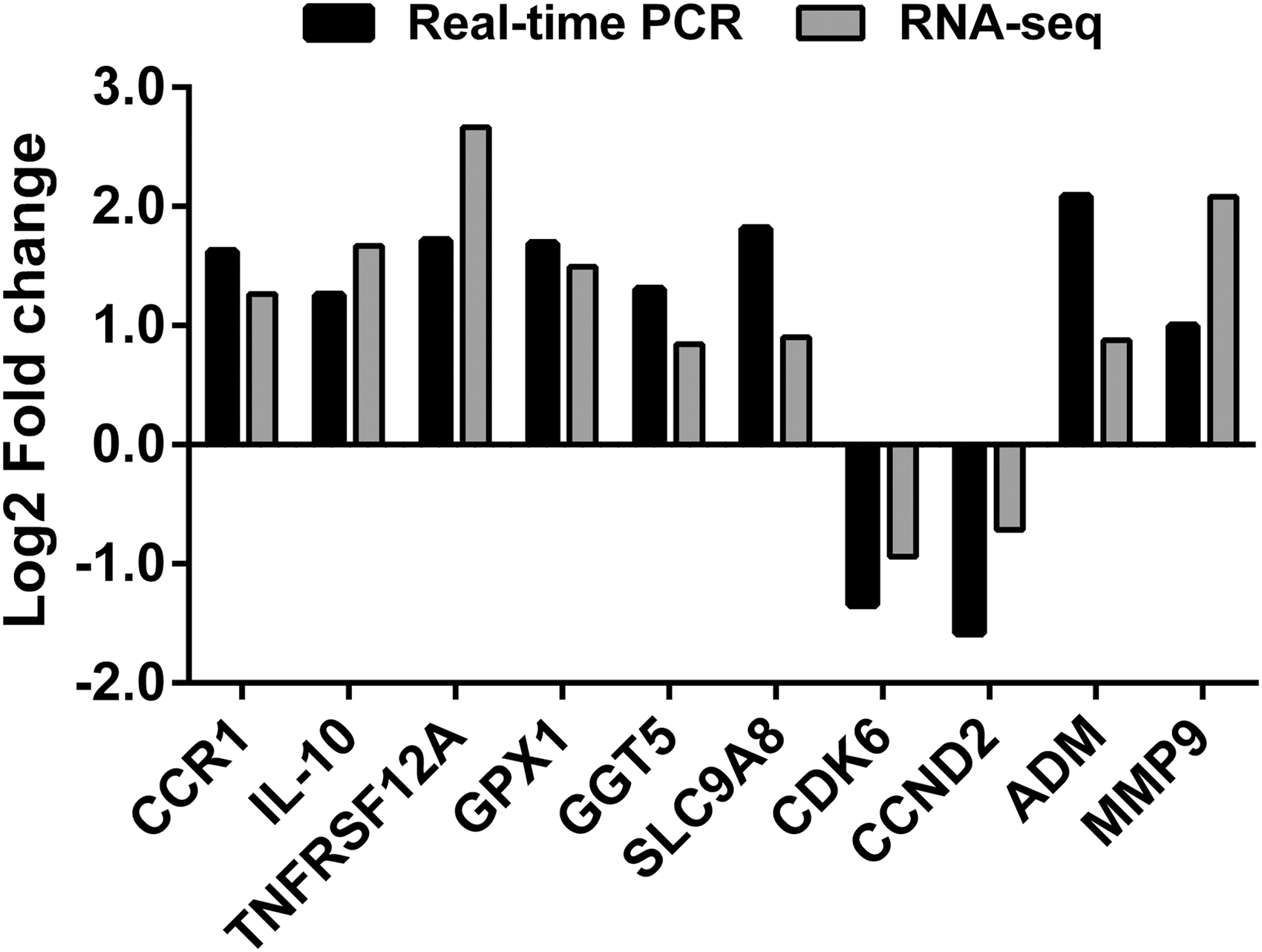
Comparison of RNA-sequencing and real-time PCR results of selected DEGs. Real-time PCR was performed on PBMC collected from 30 healthy volunteers and 30 patients within 3 h following ICH. DEG, differentially expressed gene; PCR, polymerase chain reaction.
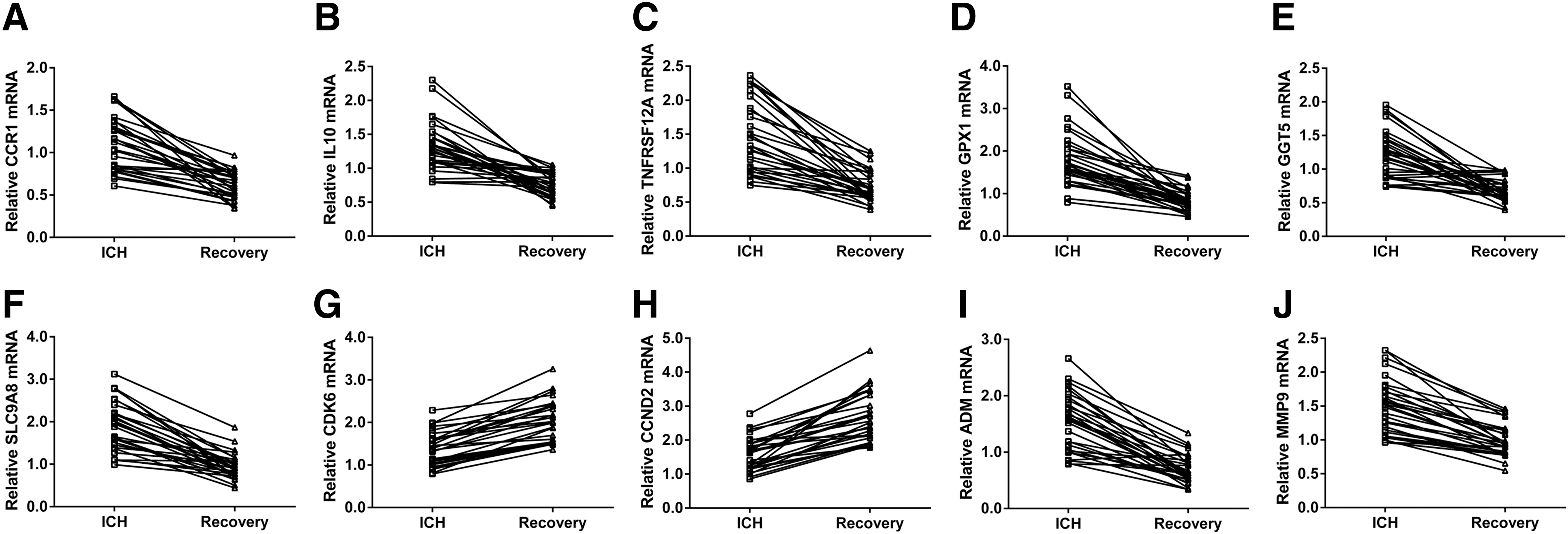
The RNA levels of selected DEGs
Discussion
Previous studies have touched on the molecular changing of peripheral blood samples during stroke by using microarray (Tang et al., 2001; Moore et al., 2005; Barr et al., 2010; Stamova et al., 2010) or protein assays (Whiteley et al., 2009; Saenger and Christenson, 2010). By microarray analyses, distinct gene expression profiles were revealed for stroke versus control subjects and probe sets were found to predict stroke (Moore et al., 2005; Barr et al., 2010; Stamova et al., 2010). In this study, we conducted RNA-sequencing for PBMCs from four ICH patients and four healthy volunteers. We identified 4040 DEGs (Supplementary Tables S1 and S2), which is several times of those identified by previous methods on stroke. Several DEGs have been reported by other groups, including IL10 (Strle et al., 2001), TNFRSF12A (Inta et al., 2008), and AQP9 (Zeng et al., 2012). Some DEGs have been identified in previous expression profiling studies on stroke (Supplementary Table S5), such as GAS7, CD14, TLR2, CYBA, ADM (Moore et al., 2005), S100A12 (Barr et al., 2010), and MMP9 (Tang et al., 2001; Power et al., 2003; Barr et al., 2010). Besides, we also identified many novel DEGs. These data suggest that the RNA-sequencing-based transcriptome analysis is a powerful tool to study expression profiling.
Functional analyses suggested that 4040 DEGs were mostly enriched in the 61 Go terms and 40 KEGG pathways, which provided important information for understanding the molecular mechanisms of ICH pathogenesis. During the course of ICH, blood rapidly enters the brain parenchyma, which results in the hindrance of the oxygen and glucose transport in the cells of the hematoma, and the neuronal apoptosis and necrosis (Felberg et al., 2002; Qureshi et al., 2003; Wasserman and Schlichter, 2007). Here as expected, the DEGs were found enriched in the following GO terms, including cell apoptosis and autophagy.
It is well known that inflammation accounts for the progression of stroke (del Zoppo and Hallenbeck, 2000; Gong et al., 2000; Zhang et al., 2006; Wang et al., 2007). Here, GO term “inflammatory response,” KEGG chemokine signaling, and cytokine–cytokine receptor interaction were enriched in the PBMCs of ICH patients. CCR1 (Carmichael et al., 2008), IL10 (an anti-inflammatory cytokine) (Pelidou et al., 1999; Strle et al., 2001) and TNFRSF12A (Inta et al., 2008) is found upregulated in experimental stroke or stroke patients. Consistent with these previous studies, CCR1, IL10, and TNFRSF12A were upregulated in PBMCs of ICH patients as indicated by RNA-sequencing (Supplementary Table S1) and real-time PCR analysis (Fig. 3). Further, CCR1, IL10, and TNFRSF12A were significantly decreased at 6 months after ICH (Fig. 4). These findings confirmed the involvement of inflammatory response in the pathogenesis and recovery of ICH.
Oxidative stress plays an important role in neurodegenerative diseases of the central nervous system and ICH (Hu et al., 2016). Here, KEGG oxidative phosphorylation and glutathione metabolism processes were enriched in the PBMCs of ICH patients. Following ICH, increased reactive oxygen species contributes to MMP9 activation and neuronal cell death (Tejima et al., 2007; Katsu et al., 2010; Wu et al., 2011). MMP inhibition can reduce infarct size, brain edema, and hemorrhage (Pfefferkorn and Rosenberg, 2003). Polymorphism in the GPX1 gene, a key enzyme of the antioxidant system, was shown to be related to ICH (Pera et al., 2008). Serum gamma-Glutamyltransferase has been regarded as a risk factor of ischemic stroke (Jousilahti et al., 2000; Emdin et al., 2002). In line with these previous studies, MMP9, GPX1, and GGT5 were upregulated in PBMCs of ICH patients as indicated by RNA-sequencing (Supplementary Table S1) and real-time PCR analysis (Fig. 3). Further, MMP9, GPX1, and GGT5 were significantly decreased at 6 months after ICH (Fig. 4). These findings confirmed the importance of oxidative stress in the pathogenesis and recovery of ICH.
Conclusion
In this study, we utilized next generation sequencing platform to comprehensively characterize PBMCs transcriptomes in response to acute ICH. We also identified GO terms and KEGG pathways strongly associated with ICH by bioinformatics analysis. This study provides a basis for the understanding of the molecular mechanisms of ICH pathogenesis at system-wide levels and gives useful information for diagnosis and management of ICH, although the specificity of gene signatures for ICH needs to be further determined and the clinical potential requires further exploration.
Footnotes
Acknowledgments
This work was supported by the Initial Project for PhD of Hubei University of Medicine [2015QDJZR03], the Open Project of Hubei Key Laboratory of Wudang Local Chinese Medicine Research (Hubei University of Medicine) [WDCM005], and the Young Scientist Innovation Team Project of Hubei College [T201510].
Disclosure Statement
All authors declare that there are no conflicts of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.