
Abstract
microRNAs (miRNAs) have been proved to be involved in many events of tumor development and progression, including cell proliferation, cell apoptosis, and cell cycle arrest. However, the potential role of miR-144-3p in pancreatic cancer (PC) remains elusive. In this study, we demonstrated that miR-144-3p was decreased in PC tissues and PANC-1 cells, whereas proline-rich protein 11 (PRR11) was remarkably increased. miR-144-3p mimics were discovered to inhibit cell proliferation by arresting cells at the S-phase of the cell cycle, and inducing cell apoptosis in PANC-1 cells. The effects of miR-144-3p on cell proliferation and cell apoptosis were reversed after treatment with the miR-144-3p inhibitor. Furthermore, a luciferase activity assay indicated that miR-144-3p directly targeted PRR11 3′-UTR. Moreover, transfection with miR-144-3p mimics inhibited the expression of PRR11. miR-144-3p mimics also upregulated the expression of p-JNK and p-p38, whereas they downregulated the expression of p-ERK. The effects of miR-144-3p on mitogen-activated protein kinase pathway proteins were reversed by the miR-144-3p inhibitor. PRR11 overexpression attenuated the effect of miR-144-3p mimics on cell apoptosis and cell cycle arrest. The expression of caspase-3 was decreased by enhanced PRR11. In summary, our findings indicated that miR-144-3p induced cell cycle arrest and apoptosis in PC by targeting PRR11. Therefore, the targeting of miR-144-3p could serve as a potential therapeutic strategy for the treatment of PC.
Introduction
P
microRNAs (mRNAs) as small noncoding RNAs of ∼18–25 nucleotides are involved in the regulation of cancer development and progression in various types of cancer, acting as either oncogenes or tumor suppressor genes (Giordano and Columbano, 2013). The basic mechanism of miRNAs action is that miRNAs can bind to the 3′-UTR of target mRNAs, resulting in translational repression or target mRNA cleavage (Iwakawa and Tomari, 2015). Growing evidence suggests that miRNAs play an important role in various biological processes, including cell proliferation, apoptosis, and differentiation (Bartel, 2004).
Microarrays have identified a number of miRNAs, such as miR-132, miR-34, miR-506, and miR-21 (Dillhoff et al., 2008; Wang et al., 2010; Zhang et al., 2011; Li et al., 2016), that are up- or downregulated in PC. MiR-144, including miR-144-3p, is a well-known onco-miRNA and has been reported to be deregulated in other types of cancer, such as gastric cancer, breast cancer, and colorectal cancer (Liu et al., 2015; Yu et al., 2015). However, the exact role of miR-144-3p in PC has not yet been revealed. Nevertheless, whether and how miR-144-3p is involved in the pathogenesis of PC is the key point of this study.
Dysregulation of cell cycle components promotes tumor formation, and, therefore, the proteins controlling cell cycle progression are potential targets for anticancer strategies (Williams and Stoeber, 2012). Proline-rich protein 11 (PRR11) was identified as a novel gene and was functionally characterized by Ji et al. (2013), who discovered that PRR11 exerts a vital role in both cell cycle progression and tumorigenesis (Ji et al., 2013).
PRR11, a candidate oncogene, has been implicated in the regulation of cell cycle progression as well as in lung cancer development (Ji et al., 2013). The silencing of PRR11 induces S-phase arrest, inhibiting cell viability and tumorigenic potential (Ji et al., 2013). In addition, in silico analysis has suggested that high expression of PRR11 is significantly associated with poor prognosis in lung cancer patients (Ji et al., 2013). PRR11 might serve as a new potential target in the treatment of lung cancer. Considerable studies have shown that PRR11 was overexpressed in gastric, pancreatic, and colorectal cancers and hilar cholangiocarcinoma (Chen et al., 2015b; Song et al., 2015; Tao et al., 2015). However, whether and how PRR11 is involved in PC is not very clear at present.
Mitogen-activated protein kinases (MAPKs), such as the protein Ser/Thr kinases, are important regulators of cell growth and survival in physiological and pathological processes (Johnson and Lapadat, 2002). Abnormal expression of MAPK signaling proteins exerts an important influence on the development and progression of various cancers, as well as in determining response to cancer treatments (Santarpia et al., 2012). Three MAPK genes (PyERK, PyJNK, and Pyp38) are closely related to different types of cancer (Lange-Carter et al., 1994; Mulholland et al., 2012; Ahronian et al., 2015). The open reading frames of PyERK, PyJNK, and Pyp38 are 1104, 1227, and 1104 bp, encoding 367, 408, and 367 amino acids, respectively. This article will further explore whether the MAPK signaling pathway participates in the development process of PC.
Our study demonstrated that miR-144-3p was decreased in PC tissues and cells. In addition, upregulation of miR-144-3p triggered cell cycle arrest and enhanced cell apoptosis in PANC-1 cells by targeting PRR11. We found that miR-144-3p acts as a novel tumor suppressor in PC. Our findings suggest that the tumor-suppressive function of miR-144-3p is mediated, in part, by PRR11 via the MAPK signaling pathway.
Materials and Methods
Study subject and sample collection
A total of 40 PC and adjacent noncancerous tissue samples (each for 20 samples) were obtained from patients who had undergone surgical treatment for PC in the First Affiliated Hospital of Xinxiang Medical University. The patients were diagnosed, and all tissue samples were taken independently by two experienced pathologists from the First Affiliated Hospital of Xinxiang Medical University, according to the Cancer Staging Manual published by the American Joint Committee on Cancer. The present study was approved by the First Affiliated Hospital of Xinxiang Medical University, and informed consent was obtained from the patients before sample collection, conforming to the Declaration of Helsinki and local legislation. Written informed consent was obtained from all patients before the start of the study.
Cell lines and culture
HPDE6-C7 cells (a normal human pancreatic duct epithelial cell line) and PANC-1 cells (American Type Culture Collection, Manassas, VA) were cultured in mycoplasma-free Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum, 100 IU/mL penicillin sodium, and 100 μg/mL streptomycin sulfate at 37°C in a 5% carbon dioxide atmosphere. All other chemicals and solvents were commercially available.
Cell transfection
A pcDNA3.1-PRR11 plasmid was constructed by inserting a cDNA fragment retro-transcribed from the coding sequence without a 3′-UTR. When PANC-1 cells reached 80% confluence, the miR-144-3p mimics or miR-144-3p inhibitor (GenePharma, Shanghai, China), pcDNA3.1-PRR11 or empty vector were transfected into PANC-1 cells by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. In the assay, three parallel wells were made.
Luciferase reporter assay
PRR11 3′-UTR was amplified from PANC-1 cells with specific primers and was then cloned into a pMiR-Report vector system (Ambion Inc., Austin, TX). A mutated 3′-UTR of PRR11 was introduced into the potential miR-144-3p binding site by using a two-step polymerase chain reaction (PCR) approach. PANC-1 cells were co-transfected with the reporter vectors containing the wild type or mutant of PRR11 3′-UTR and miR-144-3p mimic or miR-144-3p inhibitor. After 48 h, luciferase activity was measured by using a dual-luciferase reporter assay system (Promega, Madison, WI). Experiments were repeated three times in triplicate.
Quantitative reverse transcription-PCR analysis
Total RNA was extracted by using TRIzol reagent (Invitrogen) and miRNeasy kits (Qiagen, Hilden, Germany) according to the manufacturer's protocol. The miRNA was converted to cDNA by using a PrimeScript RT reagent kit (TaKaRa, Tokyo, Japan) according to the manufacturer's protocol. miR-144-3p and PRR11 were investigated by using an miRVana real-time PCR miRNA Detection Kit and real-time PCR Primer Sets. The primers used for miR-144-3p and PRR11 are as follows: miR-144-3p: 5′-GCGCTACAGTATAGATGATGTAC-3′. PRR11: forward primer: 5′-CGTATCTGCCACCGAGAACTT-3′, reverse primer: 5′-GAGATGGTCTTCAGTGCTTCCT-3′. GAPDH: forward primer: 5′-TGACTTCAACAGCGACACCCA-3′, reverse: 5′-CACCCTGTTGCTGTAGCCAAA-3′. Each sample was analyzed in triplicate.
Western blot analysis
The Bradford assay (Bio-Rad Laboratories, Hercules, CA) was used to quantify protein concentrations. All proteins from tissues were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, blotted, and probed with primary antibodies against PRR11(1:1000, Invitrogen), CyclinD1, CDC25, p21, Caspase-3, JNK, p-JNK, p38, p-p38, ERK, p-ERK, and β-actin (1:1000, all from Santa Cruz Biotechnology Inc., CA). Horseradish peroxidase-conjugated anti-rabbit secondary antibodies (1:10000, Santa Cruz Biotechnology Inc.) were used and visualized by using an enhanced chemiluminescence kit (Thermo Scientific, Waltham, MA). The density of each band was quantified with Image J 1.42q software (National Institutes of Health, Bethesda, MD). Data were normalized to β-actin. Each sample was analyzed in triplicate.
Cell viability assay
Cell vitality was analyzed by using the 3-(4, 5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide assay. Briefly, cells were treated with a total of 20 mL of the MTT solution (5 mg/mL; Sigma, St. Louis, MO) and subsequently incubated with 150 μL of dimethyl sulfoxide for 5 min at room temperature. Finally, optical density was determined at 570 nm. In the assay, five parallel wells were made.
Flow cytometry analysis
Cell apoptosis of PANC-1 cells was detected with the flow cytometry method (FCM) by using an Annexin V-Propidium Iodide (PI) Apoptosis Detection Kit (Abcam, Cambridge, United Kingdom). Briefly, the cells were collected, washed with phosphate-buffered saline (PBS), and suspended in 500 mL of binding buffer. The cells were incubated with Annexin V at room temperature for 10 min and were then stained by PI. The percentage of apoptotic cells was analyzed by using FCM.
PANC-1 cells were harvested, collected, resuspended in PBS, permeabilized in 0.1% Triton X-100, and stained with 1 μg/mL 4′,6-diamidino-2-phenylindole for 30 min. Multicycle software version 2.5 (Phoenix Flow Systems, San Diego, CA) was used for cell cycle analysis. Each treatment was carried out in triplicate.
Statistical analysis
All statistical values were represented as the mean ± standard deviation, and they were analyzed with a one-way repeated-measures analysis of variance and Student's t-test. All statistical parameters were calculated by using SPSS 20.0 software. Differences with p < 0.05 were considered statistically significant.
Results
Expression of miR-144-3p was downregulated and PRR11 was upregulated in PC tissues and PANC-1 cells
In the present study, we evaluated the levels of miR-144-3p in PC tissues and PANC-1 cells by using an RT-PCR assay. As shown in Figure 1A and D, the levels of miR-144-3p in PC tissues and PANC-1 cells were significantly decreased in comparison with the levels in normal tissues and HPDE6-C7 cells, respectively (p < 0.05). In addition, the RT-PCR assay indicated that the mRNA levels of PRR11 were remarkably upregulated in PC tissues and PANC-1 cells compared with normal tissues and HPDE6-C7 cells (Fig. 1B, E) (p < 0.05). Similarly, compared with the normal group, Western blot analysis demonstrated that the protein expression of PRR11 was also upregulated in PC tissues and PANC-1 cells (Fig. 1C, F) (p < 0.05).
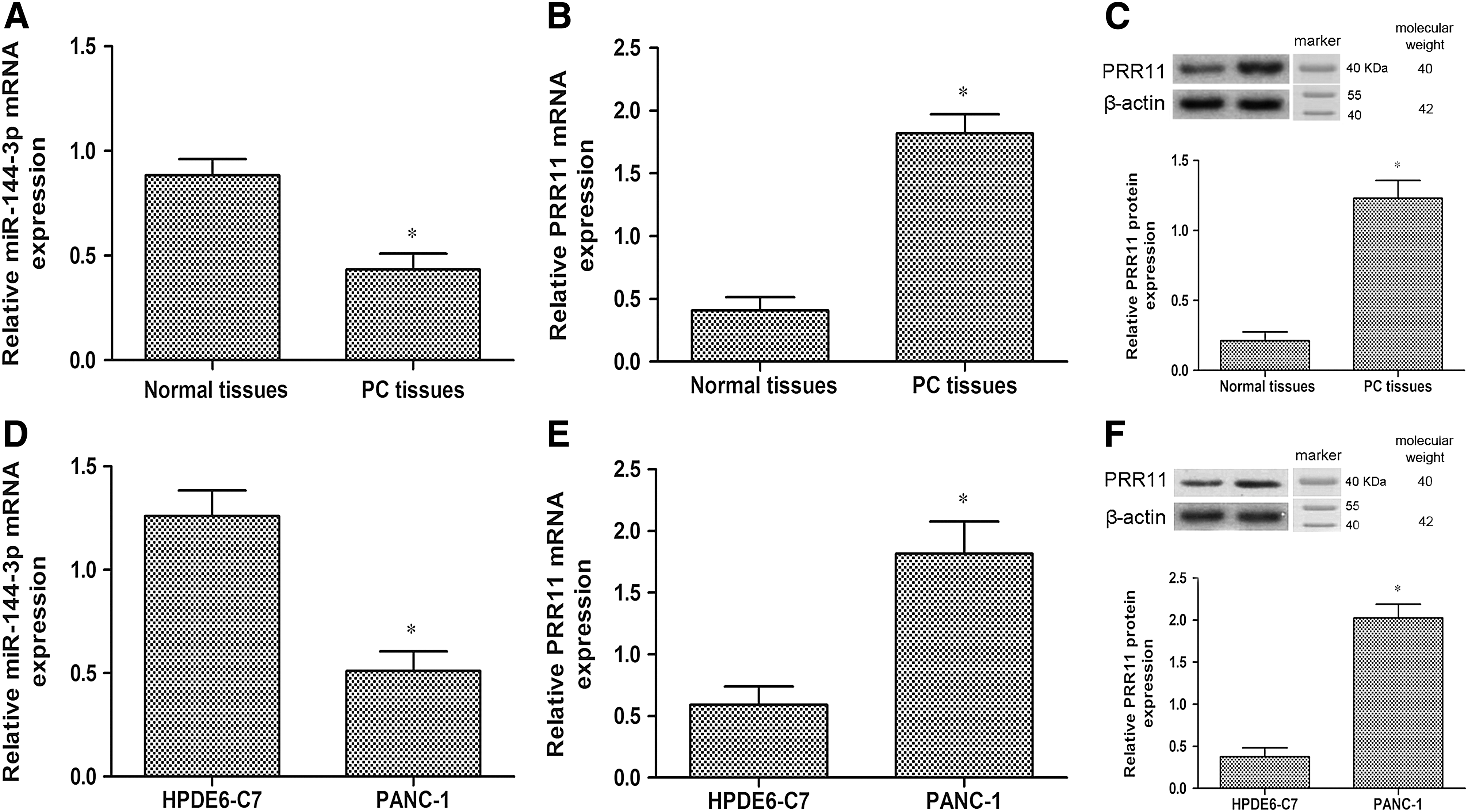
Expression of miR-144-3p and PRR11 in normal tissues and HPDE6-C7 cells, as well as PC tissues and PANC-1 cells.
miR-144-3p mimics induced cell cycle arrest and apoptosis in PANC-1 cells
Since miR-144-3p was significantly downregulated in PC, we speculated that miR-144-3p might be involved in the development of PC. To evaluate the effect of miR-144-3p on cell proliferation, cell apoptosis, and cell cycle arrest in PC, miR-144-3p mimics and inhibitor were transfected into PANC-1 cells. As demonstrated in Figure 2A, an MTT assay indicated that miR-144-3p mimics inhibited the growth of PANC-1 cells, whereas the inhibitor enhanced the growth of PANC-1cells (p < 0.05). The percentage of cells undergoing apoptosis was measured by using flow cytometry analysis. The results indicated that miR-144-3p mimics induced PANC-1 cell apoptosis (Fig. 2B) (p < 0.05). Conversely, compared with PANC-1 cells, miR-144-3p inhibitor halted cell apoptosis (p < 0.05).

Effects of miR-144-3p transfection on cell cycle arrest and cell apoptosis in PANC-1 cells.
We further examined the effects of miR-144-3p in modulating the cell cycle in PANC-1 cells. Compared with the PANC-1 cells, miR-144-3p mimics promoted PANC-1 cell cycle arrest at the S-phase, as well as restrained PANC-1 cell transit to the G2/M phase (Fig. 2C) (p < 0.05). In contrast, miR-144-3p inhibitor caused a reduction in the proportion of cells in the S-phase and an increment of cells in the G2 phase compared with the PANC-1 cells (p < 0.05). Western blot analysis also indicated that miR-144-3p mimics inhibited the expression of CyclinD1 and Cdc25A in PANC-1 cells, but increased p21 expression (Fig. 2C) (p < 0.05). The effects of miR-144-3p mimics on cell cycle proteins were reversed by the miR-144-3p inhibitor (p < 0.05).
miR-144-3p targets PRR11 in PANC-1 cells
PRR11, which is closely associated with PC development, was found to be a potential target of miR-144-3p by targetscan prediction software (Fig. 3A). To confirm whether miR-144-3p targeted PRR11 3′-UTR, a luciferase activity assay was carried out. The results showed that the relative luciferase activity was significantly decreased in PANC-1 cells transfected with wild-type PRR11 3′-UTR and miR-144-3p mimics (p < 0.05).
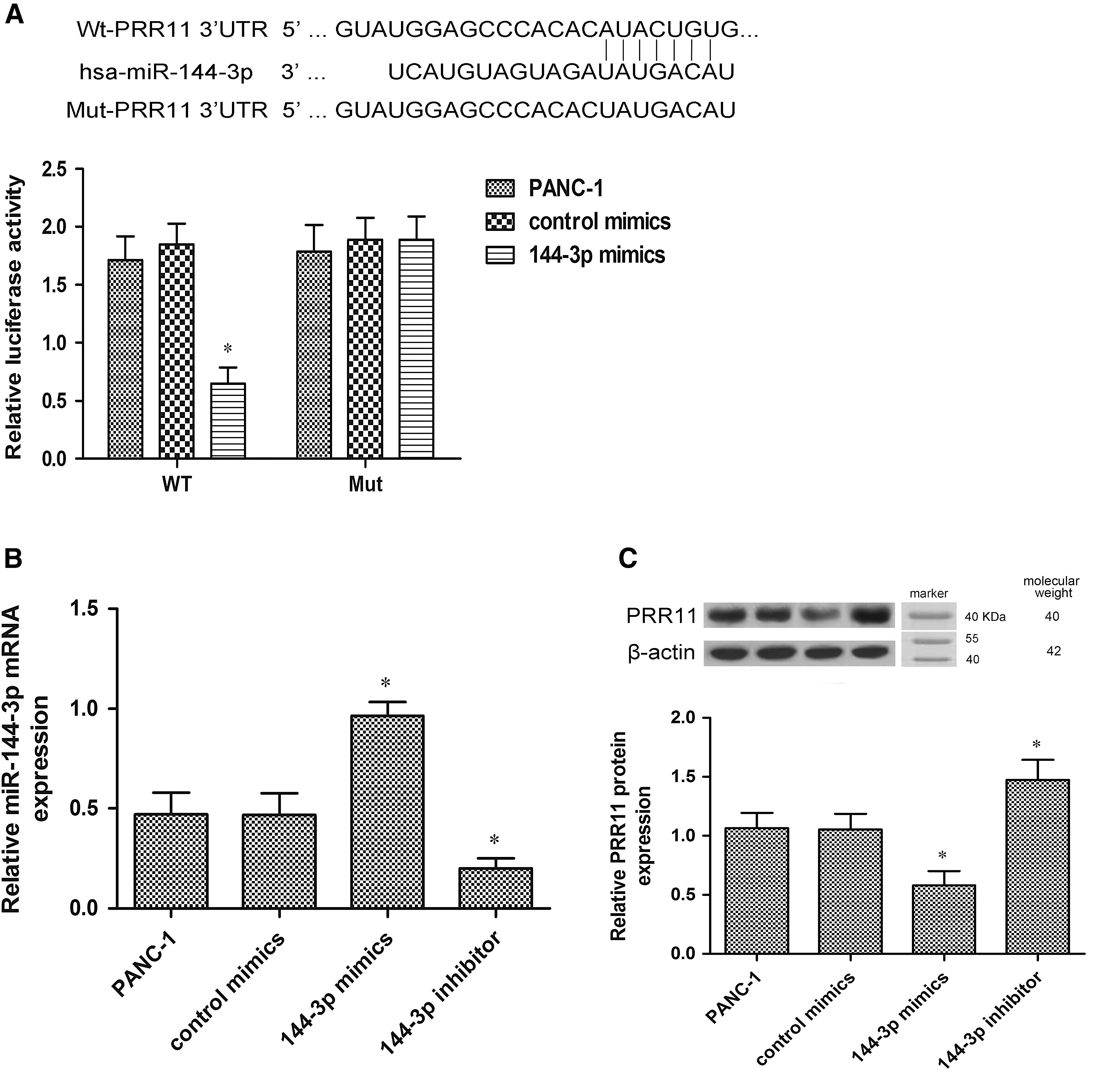
miR-144-3p targets PRR11 in PANC-1 cells.
As shown in Figure 3B, the RT-PCR assay demonstrated that the miR-144-3p levels were significantly increased in PANC-1 cells after miR-144-3p mimic transfection, and the miR-144-3p levels were decreased after miR-144-3p inhibitor transfection (p < 0.05). Western blot analysis indicated that PRR11 protein expression was downregulated in PANC-1 cells after miR-144-3p mimic transfection, and the expression was upregulated after miR-144-3p inhibitor transfection (Fig. 3D) (p < 0.05).
PRR11 overexpression reversed the miR-144-3p-mediated effect on PANC-1 cells
As shown in Figure 4A, the expression of p-JNK and p-38 was increased, whereas the expression of p-ERK was decreased by miR-144-3p mimics (p < 0.05). And the opposite results were caused by the miR-144-3p inhibitor (Fig. 4A, B) (p < 0.05).
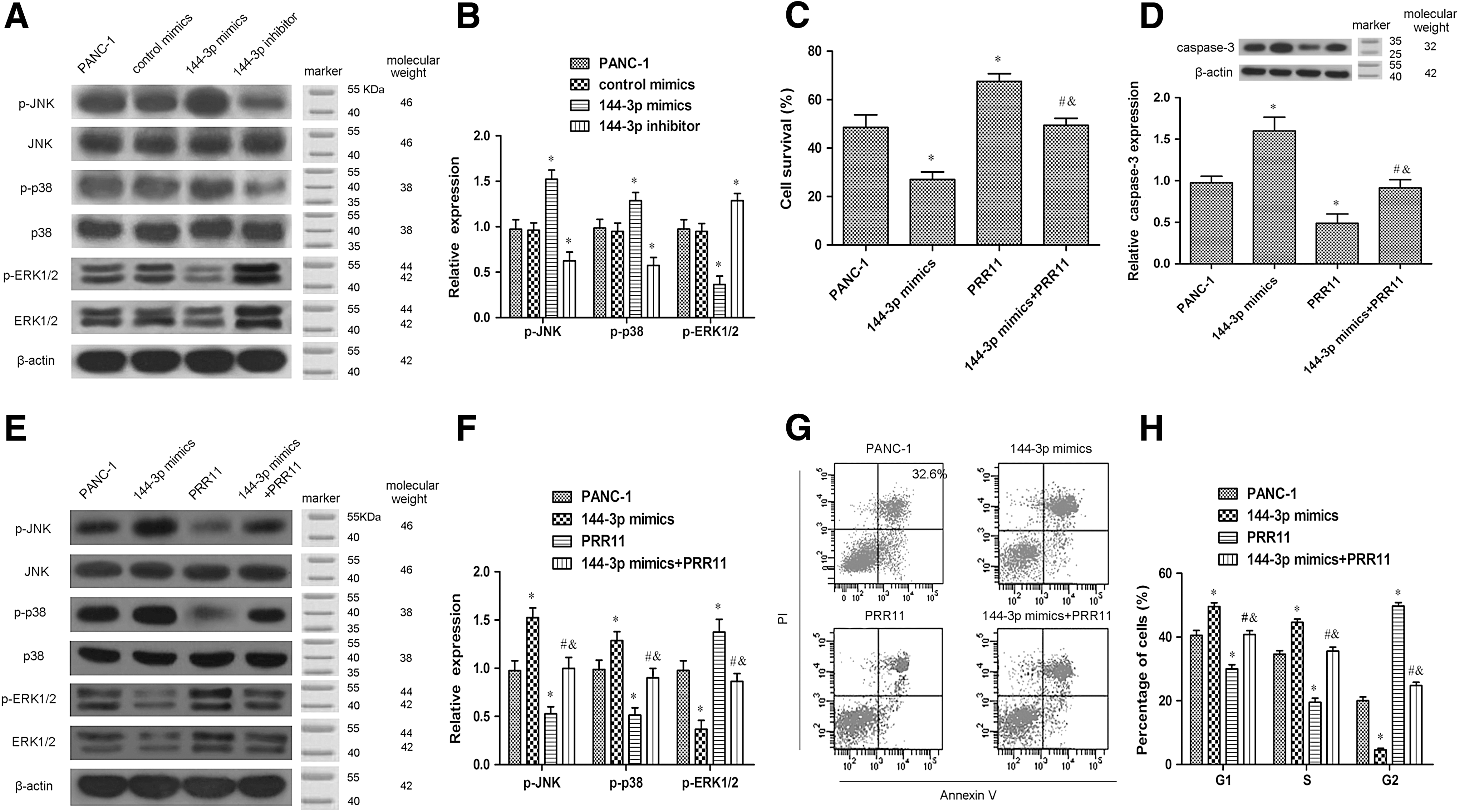
PRR11 overexpression reversed the miR-144-3p-mediated effect on PANC-1 cells.
To confirm whether PRR11 was involved in the effect of miR-144-3p during PC development, PRR11 was overexpressed in the PANC-1 cells transfected with or without miR-144-3p mimics. As shown in Figure 4C, we observed that enhanced PRR11 significantly increased the PANC-1 cell survival rate, which was blocked by miR-144-3p treatment (p < 0.05). We also found that the expression of caspase-3 was upregulated in PANC-1 cells after transfection with miR-144-3p mimics (Fig. 4D) (p < 0.05). In contrast, transfection with PRR11 overexpression intensely reversed the upregulated caspase-3 in the PANC-1 cells (Fig. 4D) (p < 0.05).
The expression of p-JNK and p-38 was decreased, whereas the expression of p-ERK was increased in PANC-1 cells after transfection with PRR11 overexpression (Fig. 4E, F) (p < 0.05). Similarly, the effects of PRR11 on the expression of p-JNK, p-38, and p-ERK were restored by miR-144-3p mimics (Fig. 4 E, F) (p < 0.05). As shown in Figure 4G and H, the results revealed that upregulation of PRR11, nevertheless, distinctly inhibited PANC-1 cell apoptosis and cell cycle arrest, and these changes were abolished by co-transfected miR-144-3p mimics (p < 0.05).
Discussion
At the time of PC diagnosis, most patients are determined as having unresectable disease (He et al., 2014; Herreros-Villanueva et al., 2014; Sun et al., 2016). Considerable efforts have been made to develop novel therapeutic treatments for this disease, but progress on this issue is limited (Wang et al., 2014). Seeking the appropriate miRNAs and tapping into the excellent potential of miRNAs will have great beneficial impact on the treatment of PC.
miR-144 plays key roles in the occurrence, development, invasion, and metastasis of some cancers (Keller et al., 2014; Zhang et al., 2015b). Previous studies suggest that anti-tumor or oncogenic effects of miR-144-3p vary depending on cancer type (Iwaya et al., 2012; Guo et al., 2013; Zha et al., 2013). It has been reported that miR-144-3p downregulation increases bladder cancer cell proliferation by targeting EZH2 and regulating Wnt signaling (Guo et al., 2013). miR-144 induced apoptosis in lung cancer cells by targeting TIGAR (Chen et al., 2015a). miR-144-3p inhibited cell proliferation and induced S/G2 cell cycle arrest in renal cell carcinoma (Xiang et al., 2016). Studies of miR-144-3p in PC have remained unclear. Therefore, we will focus on the possible functions of miR-144-3p in PC.
In the present study, we confirmed that miR-144-3p was downregulated in PC tissues and cells, whereas PRR11 expression was significantly upregulated. Further, we found that overexpression of miR-144-3p suppressed cell proliferation and induced cell apoptosis in PANC-1 cells. The amount of PANC-1 cells started to increase in the S-phase, and it was retained until just before mitotic telophase. Meanwhile, the miR-144-3p inhibitor reversed the effects of miR-144-3p mimics. These results show that miR-144-3p plays an inhibitory role in PC progression. Further studies are necessary to elucidate the mechanisms underlying the effects of miR-144-3p.
PRR11 is a relatively novel protein molecule that may play a role in cancer pathogenesis, and there is currently only a small number of articles describing the potential roles of PRR11 in lung cancer, gastric cancer, and hilar cholangiocarcinoma (Ji et al., 2013; Chen et al., 2015b; Song et al., 2015). In prior studies, PRR11 was suggested to be a negative effector in the cell cycle regulation of some tumors (Medina-Aguilar et al., 2016). Silencing of PRR11 expression resulted in significant S-phase arrest in lung cancer cells, further supporting the conclusion that cell cycle progression was inhibited after PRR11 depletion (Mi et al., 2015). In vitro and in vivo studies revealed that knock-down of PRR11 inhibited gastric cancer cell proliferation and colony-forming ability (Song et al., 2015).
Consistent with those observations, siRNA-mediated knockdown of PRR11 led to a significant cell cycle retardation in the late S-phase in H1299 cells. Surprisingly, however, it is inconsistent with previous studies that forced that the expression of PRR11 promoted the premature chromatin condensation (PCC), inhibited cellular proliferation, and caused the subsequent cell death (Zhang et al., 2015a). The authors suspected that this is because aberrant PRR11 overexpression probably triggered genomic instability and/or aneuploidy through the induction of PCC in cancerous cells (Zhang et al., 2015a).
PRR11 played multiple roles in cancer progression, indicating that PRR11 in combination with other tumor-related proteins exerts its carcinogenesis. But beyond that, new research shows that the gene pair PRR11 and SKA2 shared a bidirectional promoter and contributed to lung cancer development, which was negatively regulated by p53 through NF-Y in lung cancer cells (Wang et al., 2015, 2017). However, little is known about its role in PC. Here, we found that PRR11 was overexpressed at both mRNA and protein levels in PC tissues and cells, and we also identified PRR11 as a direct target of miR-144-3p. miR-144-3p mimics directly suppressed PRR11 expression. These data suggest that PRR11 plays a crucial role in the progression of PC. However, the precise molecular mechanisms need to be further clarified.
To investigate the exact mechanism of miR-144-3p in PANC-1 cells by targeting PRR11, we focused on the MAPK signaling pathway and its correlation with PC. miR-144-3p mimics upregulated p-JNK and p-p38 expression in PANC-1 cells, but they downregulated p-ERK expression. The results were consistent with previous results regarding the MAPK pathway in PC (Boucher et al., 2000; Matsuda et al., 2002).
Aberrant PRR11expression leads to dysregulation of several pathways and genes involved in tumorigenesis (Ji et al., 2013). Overexpression of PRR11 in PANC-1 cells may provide more evidence. As shown by the results, PRR11 overexpression abolished growth retardation in PANC-1 cells. The results strongly indicated that PRR11 regulates cell cycle progression, especially S-phase progression in PC. The expression of p-JNK and p-p38 was downregulated in PANC-1 cells after PRR11 overexpression, and PRR11 overexpression upregulated p-ERK expression. The effects of PRR11 overexpression were reversed by miR-144-3p. In addition, miR-144-3p mimics enhanced caspase-3 expression and cell apoptosis, inhibited cell proliferation, whereas the increase was reversed by PRR11 overexpression.
Taken together, our data clearly indicated that miR-144-3p may target PRR11 to induce PANC-1 cell cycle arrest and apoptosis via the MAPK signaling pathway, supporting its utility and a possible therapeutic target of PC and a promising diagnostic biomarker.
Conclusion
Our data demonstrate the miR-144-3p is an essential mediator of PC cell cycle arrest and apoptosis, thus offering a new target for the development of therapeutic agents against PC.
Footnotes
Disclosure Statement
No competing financial interests exist.