
Abstract
Herpes simplex virus type 1 (HSV-1) is a highly prevalent human neurotropic pathogen. HSV-1 infection is associated with a variety of diseases ranging from benign orolabial lesions to more serious and even life-threatening conditions such as herpes simplex keratitis and herpes simplex encephalitis (HSE). HSE is a rare occurrence among healthy adult individuals, but newborns are a particularly susceptible population. Type I IFN signaling has been identified as a crucial component of the innate immune response to the control of HSV-1 infection. In this study, we review the contribution of the type I IFN response to controlling HSV-1 infection, and differences in the early host response between adults and newborns that may contribute to the increased susceptibility to infection and central nervous system disease in newborns.
Introduction
H
HSV-1 has been traditionally associated with lesions of the oral mucosa, but the number of HSV-1-associated genital outbreaks is on the rise (Xu et al., 2006). In rare cases, HSV-1 infection can develop into more serious and even life-threatening conditions such as herpes simplex keratitis, herpetic hepatitis, and herpes simplex encephalitis (HSE) (James and Kimberlin, 2015). Newborns are particularly susceptible to developing these more severe manifestations of HSV disease following primary infection during birth (Corey and Wald, 2009). Unlike adults, in whom infection is often asymptomatic, ∼50% of infected newborns will develop disseminated disease or encephalitis (Corey and Wald, 2009).
Different components of the immune response, both innate and adaptive, are involved in controlling HSV-1 infection. Adaptive immune mechanisms, including the production of IFNγ by CD8+ T cells and neutralizing antibodies, have been found to be important in limiting viral replication (Milligan and Bernstein, 1995, 1997). Several studies have identified the innate type I IFN-mediated antiviral response as a central pathway necessary for controlling viral replication early and reducing mortality rates in murine models (Gresser et al., 1976; Zawatzky et al., 1982b; Pedersen et al., 1983). The contribution of type I IFN signaling to the neuropathogenesis of HSV has been confirmed in humans through genetic studies in children with a high susceptibility to HSV-1 infection. These studies identified mutations in different components of the type I IFN pathway that correlate with a higher incidence of HSE (Casrouge et al., 2006; Zhang et al., 2007; Pérez de Diego et al., 2010; Audry et al., 2011; Guo et al., 2011; Sancho-Shimizu et al., 2011a, 2011b; Herman et al., 2012).
However, specific mutations in components of the immune response fail to completely account for the striking difference in susceptibility to HSV-1 infection between adults and newborns. The incidence of HSV encephalitis is significantly higher in the newborn compared with the adult. This suggests that there is a strong developmental component to HSV susceptibility in the newborn. The mechanisms mediating this age-dependent susceptibility to HSV-1 infection seem to be multifactorial and remain to be fully understood. In this study, we review the type I IFN response in the context of HSV-1 infection and its contribution to increased susceptibility in the newborn.
The Type I IFN Response
The type I IFN response is initiated when host pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) in infected cells. Toll-like receptors (TLRs) and cytosolic nucleic acid sensors (RIG-1, MDA5, cGAS, and DAI) are two of the main families of PRRs. Most TLRs signal primarily through the adaptor protein MyD88, except for TLR3 whose signaling is mediated by TRIF (Takeda and Akira, 2004). Cytosolic nucleic acid sensors, on the other hand, signal primarily through the adaptor proteins MAVS and STING (Wu and Chen, 2014). Recognition of PAMPs by the PRRs and subsequent activation of adaptor proteins results in the activation of the transcription factors IRF3 (Liu et al., 2015), IRF7 (Ning et al., 2011), and NF-κB (Kawai and Akira, 2007) and their subsequent translocation to the nucleus. IRF3 and IRF7 induce transcription of type I IFNs (IFNα and IFNβ), which are secreted and act in an autocrine and paracrine manner to induce an antiviral state in cells. NF-κB induces production of proinflammatory cytokines and chemokines, which are involved in the recruitment of immune cells and induction of the inflammatory response.
The interaction of IFNs with IFN receptors leads to activation of the JAK/STAT signaling pathway. Type I IFNs signal primarily through phosphorylation of transcription factor STAT1. Phosphorylated STAT1 homodimers translocate to the nucleus and induce upregulation of several interferon-stimulated genes (ISGs) (Shuai et al., 1994). Some examples of major ISGs are protein kinase R (PKR), which phosphorylates eIF2α inhibiting cellular and viral mRNA translation (Rajesh et al., 2015), and RNase L, which degrades RNA when activated by the ISG OAS.
HSV-1 and the Type I IFN Response
Early studies with mouse strains that were either susceptible or resistant to HSV infection identified the type I IFN pathway as a significant contributor to resistance from HSV disease (Lopez, 1975), which served as the first evidence for the key role of the type I IFN response in controlling HSV-1. These studies were further supported by the observation that treatment with anti-interferon serum increased mortality in resistant mice (Gill et al., 2006). The development of mouse models with genetic ablation of specific genes that participate in the IFN response has provided more definitive evidence for the importance of the IFN response to controlling HSV infection (Leib et al., 1999; Gill et al., 2006).
Multiple components of the cellular innate immune response that ultimately lead to the production of type I IFNs have been implicated in the recognition of HSV-1 during infection. TLR-3 and TLR-9, which recognize cytosolic dsRNA and unmethylated CpG-rich dsDNA, respectively, have been found to interact with virus-derived nucleic acids and induce the production of type I IFNs (Jacquemont and Roizman, 1975; Krug et al., 2004; Malmgaard et al., 2004). Additionally, HSV is also recognized by TLR-2, which interacts with the viral envelope glycoproteins gH/gL and gB (Leoni et al., 2012).
RNA and DNA cytosolic sensors, such as RIG-I, cGAS, and DAI, have also been found to play important roles in recognition of HSV and initiation of the IFN response cascade (Ma and He, 2014). cGAS was identified as the main HSV DNA sensor in the cytoplasm necessary for type I IFN production in response to HSV-1 infection in vitro in human fibroblasts, macrophages, and dendritic cells, as well as in vivo in a murine model (Li et al., 2013). Interestingly, cGAS has also been shown to stabilize IFI16, a nuclear exogenous DNA sensor, in human foreskin fibroblasts during HSV-1 infection (Orzalli et al., 2015). IFI16 accumulates on the HSV-1 genome and evidence suggests that it suppresses viral gene expression and restricts replication in vitro (Johnson et al., 2014). Importantly, deletion of either sensor results in comparable impairment of type I IFN production in cultured cells (Orzalli et al., 2015).
The role of more downstream effectors of the IFN response pathway, such as IRF-3 and IRF-7, has been studied using single and double knockout mouse models. These studies revealed a dominant role of IRF-7 over IRF-3 in controlling infection. Yet, both are required for controlling disease progression and orchestrating the inflammatory response to the virus (Murphy et al., 2013).
As with many viruses, HSV-1 has developed multiple strategies to evade the host immune response. ICP0, a viral E3 ubiquitin ligase, inhibits IRF-3 signaling and prevents activation of the NF-κB pathway (Melroe et al., 2007; van Lint et al., 2010). It also targets TLR signaling by promoting degradation of adaptor proteins MyD88 and Mal. The immediate early protein ICP27 targets the NF-κB and IRF-3 signaling pathways (Kim et al., 2008); γ34.5, another major virulence factor, targets several pathways to counteract the host IFN response (Chou and Roizman, 1992, 1994); γ34.5 mediates the dephosphorylation of eIF2α and IKKα to reverse host shutoff of protein synthesis (He et al., 1997; Wilcox et al., 2015a), inhibits TBK1 to prevent activation of the type I IFN response (Verpooten et al., 2009), and binds to beclin-1 to inhibit autophagy (Orvedahl et al., 2007; Gobeil and Leib, 2012). The multiple strategies developed by the virus to neutralize the host type I IFN response strongly support the essential role of this pathway in controlling HSV infection (Wilcox and Longnecker, 2016).
Newborn Susceptibility to HSV-1 Infection
The incidence of HSV infection in neonates is estimated to occur in ∼1 in 3,200 births (Brown et al., 2003). Of those infected, ∼50% will survive with severe morbidity (Corey and Wald, 2009) and one-third will develop HSE (Kimberlin, 2004). This is in stark contrast with the adult population where, despite the high prevalence of infection, most individuals are asymptomatic and the incidence of HSE is only about 1 in 1,000,000 (James and Kimberlin, 2015). Importantly, immunocompromised adults, such as HIV-infected patients (Bagdades et al., 1992), individuals undergoing chemotherapy (Herget et al., 2005), or transplant recipients (Meyers et al., 1980), have an increased probability of HSV reactivation leading to HSV-related diseases. In these adult cases, HSV disease is mostly mucocutaneous and only in rare instances results in disseminated disease or infection of the central nervous system (CNS) (Wald and Corey, 2007), suggesting that an immunodeficient state or immature immune response alone does not account for the increased susceptibility to CNS disease in the newborn compared with the adult. In children, genetic studies have identified several mutations in innate signaling pathways that confer increased susceptibility of newborns to HSE (Sancho-Shimizu et al., 2011a), but these inborn errors alone do not seem to completely account for the significantly higher incidence and poor outcome of HSV infection in this age group. Taken together, these data suggest that there are not only inherent differences in the immune response between these two populations but also the developmental state of the host is likely contributing to the increased susceptibility of the newborn to HSV disease.
Recent studies have focused on identifying differences in the immune response between newborns and adults that could account for this age-dependent susceptibility (Adkins et al., 2004). Although susceptibility to different pathogens in newborns has been traditionally attributed to a lack of immunologic memory, recent studies have suggested that more intricate mechanisms might be at play. In the adaptive immune response, a bias for TH1 over a TH2 T cell response observed in newborns suggests that the immune response in this age group is geared toward preventing a proinflammatory state (Garcia et al., 2000). This is further supported by studies showing decreased production of the proinflammatory cytokines, TNF, IFNα, IFNγ, and IL-1β, in the newborn (Maródi, 2006). These differences suggest that neonatal susceptibility is, in part, due to a deficient innate immune response. However, studies showing higher levels of IL-6, IL-8, and IL-10 in human newborn monocytes support the idea of a qualitatively different rather than a dampened immune response in the newborn (Angelone et al., 2006).
Early studies in mice identified the increased susceptibility of newborns to HSV-1 even in resistant strains (Zawatzky et al., 1982b). Following intraperitoneal inoculation, newborn mice failed to activate natural killer (NK) cells and had reduced titers of IFN compared with adults (Zawatzky et al., 1982a). This provided initial evidence that a restricted IFN response in the newborn could be associated with increased susceptibility to the virus. Other studies found that viral thymidine kinase is dispensable for neurovirulence and latency only in the newborn, but not in the adult (Hay et al., 1995). Recent studies using an IFN-α/β receptor (IFNAR) KO murine model suggest that while the type I IFN response provides a protective effect in the adult following direct challenge to the brain, it is not sufficient to control HSV-1 in the newborn (Wilcox et al., 2015b). Evidence also demonstrates that targeting of the type I IFN pathway to inhibit autophagy is required to cause disease in the adult, but is dispensable in the newborn (Wilcox et al., 2015b). Interestingly, autophagy activation in the newborn is IFN independent, is not inhibited by HSV-1, and seems to have deleterious effects associated with increasing apoptotic activity in the brain. A recent study has identified differences in basal levels of expression of different components of the type I IFN pathway between neonate and adult brains (Fig. 1A) (Wilcox et al., 2016). The IFNAR and PKR, two components previously shown to be important to control HSV infection in the brain (Leib et al., 2000; Wilcox et al., 2015a), were found to have 4- to 6-fold higher levels of basal expression in the adult, while other components such as STAT1, TBK1, cGAS, and TLR3 were present in lower levels in the adult brain. Interestingly, STING, another important component of the innate immune response to HSV in the brain (Parker et al., 2015), is present at similar levels in newborn and adult brains. This indicates that multiple factors are at play in mediating the increased susceptibility to HSE and it cannot be attributed solely to a global downregulation of type I IFN response in the newborn brain.
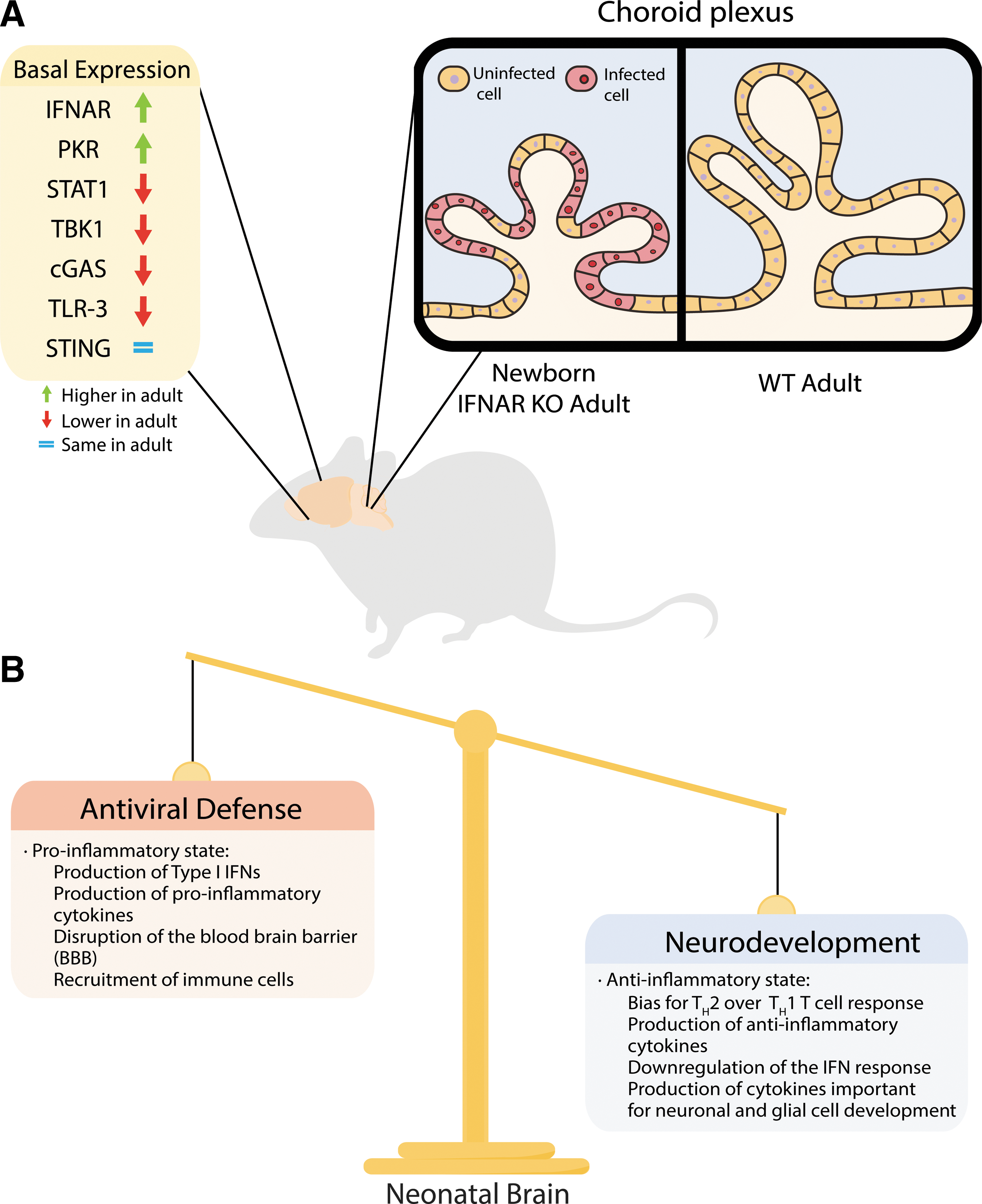
The factors responsible for the age-dependent susceptibility to HSV-1 infection observed in mice and humans may represent a careful balance between protection from pathogens and neurodevelopment.
In addition to differences in individual components of the type I IFN pathway, the IFNAR KO model also revealed important differences in the tropism of HSV-1 between the adult and the neonate following disseminated disease. The choroid plexus, a highly specialized epithelial tissue responsible for producing the cerebrospinal fluid in the brain, was found to be heavily infected in wild-type newborns, but not in adults (Wilcox et al., 2016). Interestingly, susceptibility of the adult choroid plexus was restored in IFNAR KO mice, suggesting an important role for the type I IFN response in determining viral tropism (Fig. 1A). This could have important implications for spread to the CNS following replication in the periphery as well as dissemination within the brain. These findings are further supported by previous studies showing that differences in the expression of the HSV entry receptors, HVEM and Nectin-1, between the adult and the newborn could be associated with the distinct susceptibility observed in these two populations (Kopp et al., 2013, 2014). These findings suggest that HSV infection of different cell populations of the brain might be involved in determining the different outcomes of infection between newborns and adults.
It has been hypothesized that neonatal susceptibility to different infections is the result of essential developmental processes in the newborn, such as colonization of the gut and the skin by commensal organisms, rather than an intrinsic defect in the neonatal immune system (Elahi et al., 2013). This idea suggests that susceptibility to HSV infection in the newborn could be the result of a delicate balance in the activation of the type I IFN response between restricting viral infection and inducing neurotoxicity in the developing brain (Fig. 1B). Indeed, previous studies on neonatal TLR-2-deficient mice showed increased survival following intraperitoneal injection (Kurt-Jones et al., 2004; Aravalli et al., 2005), which correlated with reduced levels of proinflammatory cytokines (Aravalli et al., 2005). Furthermore, activation of TLR-3 during development has been shown to result in impaired axonal growth and sensorimotor deficits (Cameron et al., 2007). These lines of evidence suggest that the differences in intrinsic levels of different immune components previously described (Kopp et al., 2014) could be attributed to a developmental mechanism design to prevent an inflammatory state in the newborn. The different factors contributing to the susceptibility to HSV infection in the newborn remain largely understudied and further studies are needed to understand the interplay between antiviral protection provided by the type I IFN response and its possible detrimental consequences in development.
Conclusions and Future Perspectives
The type I IFN response plays a crucial role in protecting the host from viral infection. Several in vivo and in vitro studies have demonstrated the importance of this essential component of the innate immune response to controlling HSV-1 infection. Many human and mouse models have pointed to the type I IFN response as the central pathway involved in the development of HSE. However, the marked increased susceptibility of newborns to HSE does not appear to be simply explained by a dampened IFN response. The delicate balance required to ensure proper neurodevelopment seems to have resulted in specific mechanisms that tightly regulate the IFN response in the brain and prevent neurotoxicity. Initial studies of the differences in the type I IFN response between neonates and adults have unveiled important differences in the ways HSV-1 targets this important pathway and the different mechanisms that are important for controlling the virus in these two populations. However, further studies focusing on dissecting the specific pathways that differ between neonates and adults as well as the role of different cell populations in the brain are still necessary to increase our understanding of the high prevalence of HSE in newborns.
Footnotes
Acknowledgments
Work on HSV-1 in the Longnecker Laboratory is supported by National Institutes of Health (USA) grants T32AI060523, T32GM008152, and F30AI116106 to D.W. and R01CA021776 and R01 EY023977 to R.L. The authors thank all the members of the Longnecker Laboratory, especially Nannette Susmarski and Sarah Kopp, for all their support.
Disclosure Statement
No competing financial interests exist.