
Abstract
Epigenetic dysregulation has been shown to limit functional capacity of aging hematopoietic stem cells, which may contribute to impaired outcome of hematopoietic stem cell-based therapies. The aim of our study was to gain better insight into the epigenetic profile of CD34+-enriched cell products intended for autologous CD34+ cell transplantation in patients with cardiomyopathy. We found global DNA methylation content significantly higher in immunoselected CD34+ cells compared to leukocytes in leukapheresis products (2.33 ± 1.03% vs. 1.84 ± 0.86%, p = 0.04). Global DNA hydroxymethylation content did not differ between CD34+ cells and leukocytes (p = 0.30). By measuring methylation levels of 94 stem cell transcription factors on a ready-to-use array, we identified 15 factors in which average promoter methylation was significantly different between leukocytes and CD34+ cells. The difference was highest for HOXC12 (58.18 ± 6.47% vs. 13.34 ± 24.18%, p = 0.0009) and NR2F2 (51.65 ± 25.89% vs. 7.66 ± 21.43%, p = 0.0045) genes. Our findings suggest that global DNA methylation and hydroxymethylation patterns as well as target methylation profile of selected genes in CD34+-enriched cell products do not differ significantly compared to leukapheresis products and, thus, can tell us little about the functional capacity and regenerative properties of CD34+ cells. Future studies should examine other CD34+ cell graft characteristics, which may serve as prognostic tools for autologous CD34+ cell transplantation.
Introduction
O
The variable therapeutic outcomes may depend on various factors, including patient and disease characteristics, cell graft quality, and cell graft delivery methods. Functional competence of cells used for transplantation is a prerequisite for the adequate cell graft quality. However, even in highly regenerative tissues, such as hematopoietic system, adult stem or progenitor cells do show an age-associated functional senescence with characteristic features of decreased self-renewal, reduced clonal stability, reduced homing and engraftment, and lineage commitment biased into myeloid progenitors (Chen, 2011). The mechanism underlying the decline in regenerative capacity of adult stem or progenitor cells is still poorly understood; however, a recent study indicated that loss of epigenetic regulation may drive functional attenuation of aging hematopoietic stem cells (Chambers et al., 2007).
DNA methylation is the best characterized epigenetic modification that plays an important role in gene regulation and chromatin organization. It occurs predominantly at CpG dinucleotides and is quite stable, generally maintained during cell division by DNA methyltransferases. However, during mammalian development and cell differentiation, DNA methylation is extensively reprogrammed. Methylation marks may be gained or lost at certain genes or even change genome wide. Although progress in this field has been enormous in the last decade, little is known about specific methylation changes that accompany specific developmental processes (Smith and Meissner, 2013). Given the formation of terminally differentiated hematopoietic cells is a highly hierarchical process; hematopoiesis presents a well-defined system to study DNA methylation changes through distinct developmental stages. Exploiting this notion, recent studies have described a reduction in promoter DNA methylation during mouse embryogenesis and during lineage commitment of mouse hematopoietic progenitors (Borgel et al., 2010; Ji et al., 2010). Shortly after, two independent studies showed that the same process applies to humans (Bocker et al., 2011; Calvanese et al., 2012). By using the genome-wide DNA methylation array-based approach, these studies found genes that showed strongly enriched hypomethylation in differentiated hematopoietic cells, suggesting that promoter demethylation might be an important mechanism of gene regulation during hematopoiesis (Bocker et al., 2011; Calvanese et al., 2012). Interestingly, these same sites in hematopoietic stem or progenitor cells tended to show intermediate methylation pattern that resolved to uniformity, demonstrating hypo- and hypermethylation in the opposing terminally differentiated lineages (Hodges et al., 2011).
In contrast to the DNA methylation process, active DNA methyl group removal has remained elusive until 2009 when Tahiliani et al. identified ten-eleven translocation (TET) family of enzymes, responsible for catalyzing the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC; Tahiliani et al., 2009). Apart from being an intermediate during the DNA demethylation process, 5-hmC is reported to be present at substantial levels in embryonic stem cells, primordial germ cells, and fertilized oocytes (Ruzov et al., 2011; Hahn et al., 2014). Compared to other adult tissues, 5-hmC is strongly enriched in bone marrow and brain, raising the possibility that this epigenetic mark may play a functional role in genome biology (Ruzov et al., 2011; Hahn et al., 2014). Some earlier reports suggest a model in which 5-hmC contributes to the “poised” chromatin signature found at developmentally regulated genes (Pastor et al., 2011), however, its true functional potential is only beginning to be realized and therefore needs further study.
Decline in hematopoietic stem or progenitor cell function due to epigenetic dysregulation may impair the outcome of hematopoietic stem cell-based therapies (Piccin and Morshead, 2010). The aim of our study was to gain better insight into the epigenetic profile of CD34+-enriched cell products intended for autologous CD34+ cell transplantation in patients with cardiomyopathy. We looked at the global DNA methylation and hydroxymethylation pattern of immunoselected CD34+ cells and compared that to the global DNA methylation and hydroxymethylation pattern of mainly differentiated hematopoietic cells present in corresponding leukapheresis products. In addition, we searched for candidate genes whose promoter methylation level may serve as a prognostic marker for autologous CD34+ cell transplantation in patients with cardiomyopathy.
Materials and Methods
Study population
Patients included in the study were recruited between January 1, 2014, and September 30, 2015, for autologous CD34+ cell transplantation at the Advanced Heart Failure and Transplantation Center in Ljubljana. Patient inclusion criteria were age between 18 and 70 years, diagnosis of cardiomyopathy as defined by European Society of Cardiology (ESC Working Group et al., 2013), optimal medical management for at least 6 months, left ventricular ejection fraction (LVEF) 20–50%, and New York Heart Association (NYHA) functional class heart failure II or III for at least 3 months before referral. Patients with acute multiorgan failure, history of malignant disease within 5 years, or diminished functional capacity due to noncardiac comorbidities were excluded. Informed consent was obtained from all patients before participation. The ethical approval was granted by the National Medical Ethics Committee of the Republic of Slovenia (No. 121/01/14). The clinical trial is registered at
CD34+ cell collection, immunoselection, and enumeration
Bone marrow hematopoietic cells were mobilized into peripheral blood by daily subcutaneous injections of granulocyte colony-stimulating factor (G-CSF; 10 μg/kg daily, 5 days). On the fifth day, a full blood count and peripheral blood CD34+ cell count were performed. Poor mobilizes received plerixafor 0.24 mg/kg on day 5 and continued with stem cell collection on day 6. Bone marrow-derived cells were collected by leukapheresis using Amicus cell separator (Baxter Healthcare, Chicago, IL) according to the manufacturer's instructions. Positive immunomagnetic selection of CD34+ cells from leukapheresis products was performed with the CliniMACS Plus System (Miltenyi Biotech, Bergisch Gladbach, Germany).
Before and after immunomagnetic selection, CD34+ cell number enumeration and viability were quantified by a standardized five-parameter flow cytometry method (CD45 FITC/CD34 PE staining, forward and side scatter, 7-aminoactinomycin D) according to the modified ISHAGE protocol (Sutherland et al., 2003). The addition of predefined number of counting beads to the cell suspension yielded the concentration of CD34+ cells per unit of sample volume and thus allowed for the absolute CD34+ cell count. Cell viability was determined with a 7-aminoactinomycin D. CD34+ cell recovery (absolute number of CD34+ cells after immunomagnetic selection/absolute number of CD34+ cells in leukapheresis products) and CD34+ cell purity (CD34+/leukocytes) were also calculated.
The obtained CD34+ cell products were sampled for laboratory testing and research, and a final dose of 80 × 106 CD34+ cells was sent to the Advanced Heart Failure and Transplantation Center for immediate transendocardial transplantation.
Global DNA methylation and DNA hydroxymethylation analysis
DNA was extracted from both leukapheresis and CD34+-enriched cell products using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). To obtain RNA-free genomic DNA, RNase A (100 mg/mL; Qiagen) was added to the sample during isolation process. DNA was quantified by a NanoDrop ND-1000 Spectrometer (Thermo Scientific, Wilmington, DE). Global DNA methylation and global DNA hydroxymethylation status was determined by an enzyme-linked immunosorbent assay-based method using the MethylFlash Methylated DNA Quantification Kit and MethylFlash Hydroxymethylated DNA Quantification Kit, respectively (Epigentek Group, Inc., Farmingdale, NY), according to the manufacturer's instructions. In this assay, the methylated and hydroxymethylated fractions of DNA are detected by capture and detection antibodies and then quantified colorimetrically by measuring the absorbance in a microplate reader at 450 nm. The amount of methylated and hydroxymethylated DNA is proportional to the OD measured and expressed as percentage of total DNA. Student's t-test and Pearson's correlation were applied to analyze the global DNA methylation and hydroxymethylation data.
Gene selection for target DNA methylation profiling
To select gene pathways and candidate genes for target DNA methylation profiling, we took advantage of a previously published study, which profiled DNA methylation changes during hematopoiesis by comparing the methylation maps of human embryonic stem cells (hESCs) with those of lineage-restricted progenitors and differentiated cells using HumanMethylation27 DNA Analysis BeadChip (Illumina, San Diego, CA; Calvanese et al., 2012). For the purpose of our analysis, DNA methylation data from four different cell subsets were downloaded from the NCBI Gene Expression Omnibus (GEO; Accession No. GSE30090). These included well-characterized hESCs (SHEF-1, SHEF-4, SHEF-5, SHEF-7, H7, H14, H14S9, H7S14, HS181, and I3), CD34+ cells purified from cord blood and pooled from five healthy newborns (CD34-CB), in vitro differentiated CD34+ cells (CD34-Diff), and in vitro dedifferentiated CD34+ cells, in which pluripotency was induced by viral-based ectopic expression of the reprogramming factors (CD34-induced pluripotent stem cells [iPSCs]; Calvanese et al., 2012). All CpGs with methylation values ≥70% (β ≥ 0.7) were considered hypermethylated, whereas CpGs whose equivalent signal was below 30% (β ≤ 0.3) were considered unmethylated. From 27 578 highly informative CpG sites corresponding to more than 14,000 genes, we first selected genes that were marked by hypo- and hypermethylation in all four cell subsets, hESCs, CD34-CB cells, CD34-Diff, and CD34-iPSCs. Next, we were interested which genes become demethylated and thereby possibly activated during CD34+ cell differentiation. To this end, 4 set Venn diagram was constructed with Venny 2.0.2. Computational Genomic Service (Oliveros, 2007) to identify overlapping genes that showed hypomethylation (β < 0.7) in CD34-Diff cells and hypermethylation (β ≥ 0.7) in hESCs, CD34-CB cells, and CD34-iPSCs (i.e., core set). Finally, to narrow down the number of genes for target DNA methylation screening, gene ontology (GO) analysis was performed on the core set via GSEA portal (Broad Institute; Subramanian et al., 2005). Pscan software tool (Zambelli et al., 2009) together with transcription factor-binding profiles from the JASPAR database (Mathelier et al., 2014) was used to identify enriched transcription factor binding motifs in the promoter region of the core set, −450 bp to 50 bp with respect to the gene's transcription start site.
Target DNA methylation analysis
We used restriction enzyme-based MethylScreen™ technology to profile the methylation status of target genes. Based on the results of the GO analysis, Human Stem Cell Transcription Factors EpiTect Methyl II Complete PCR Array (Qiagen) was selected, which profiles the promoter methylation level of a panel of 94 transcription factors whose association with stem cell differentiation and development has been well documented. The method used by the EpiTect Methyl II PCR System detects the remaining input DNA after cleavage with a methylation-sensitive and/or a methylation-dependent restriction enzyme by real-time PCR using primers that flank a promoter (gene) region of interest. The relative fractions of methylated and unmethylated DNA are subsequently determined by comparing the amount of each digest with that of a mock (no enzyme added) using a ΔΔCt method (Jiang et al., 2012). Student's t-test was used to analyze the difference in promoter methylation of selected genes between CD34+-enriched cell products and leukapheresis products.
Real-time reverse transcription-PCR analysis
RNA was extracted from both leukapheresis and CD34+-enriched cell products using the RNeasy Mini Kit (QIAGEN, Germantown, MD). RNA was reverse transcribed into cDNA (QuantiTect Reverse Transcription Kit; Qiagen), which was quantified by real-time PCR (ViiA 7 Real-Time PCR System; Applied Biosystems, Foster City, CA). TaqMan Gene Expression Assay (Applied Biosystems, Foster City, CA) was used to detect the expression levels of human nuclear receptor subfamily 2, group F, member 2 (NR2F2; also known as chicken ovalbumin upstream promoter transcription factor 2 [COUP-TFII]). Gene expression was quantified relative to the housekeeping gene cyclophilin (huCYC) using the ΔΔCT method. hESC line WA09 (WiCell, Madison, WI, USA) and human fibroblasts CRL-2352 (ATCC, Manassas, VA) were used as a positive and negative control for NR2F2 gene expression, respectively.
Statistical analyses
Summary statistics were used to describe participant characteristics. A p-value <0.05 was considered statistically significant. All statistical analyses were done using SPSS 22.0 software package (SPSS, Inc., Chicago, IL).
Results
Baseline patient's and cell product's characteristics
We studied 13 patients diagnosed with ischemic or nonischemic cardiomyopathy. Data on age, sex, cardiomyopathy pathogenesis, NYHA class, LVEF, left ventricular end-diastolic dimension, plasma sodium, creatinine, and N-terminal B-type natriuretic peptide of patients enrolled in the study are presented in Table 1.
Values are shown as mean ± SD, unless otherwise stated.
LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricular ejection fraction; NT-proBNP, N-terminal B-type natriuretic peptide; NYHA, New York Heart Association; SD, standard deviation.
CD34+ cell recovery (absolute number of CD34+ cells after immunomagnetic selection/absolute number of CD34+ cells in leukapheresis products), CD34+ cell purity (CD34+/leukocytes), and CD34+ cell viability of the final CD34+-enriched cell products were analyzed by flow cytometry. Mean CD34+ cell recovery was 64.66% (range 34.89–84.53%), mean CD34+ cell purity was 96.60% (range 87.50–100.00%), and mean CD34+ cell viability was 99.38% (range 98.90–99.80%), demonstrating high-quality cell grafts.
Global DNA methylation and hydroxymethylation
Based on previous studies indicating the DNA hypomethylation process taking place during hematopoietic differentiation, we compared the global 5-mC levels in mainly differentiated hematopoietic cells in leukapheresis products with the global 5-mC levels in immunoselected CD34+ cells. In accordance with CD34+ cell's more primitive character, we found global 5-mC content in CD34+-enriched cell products slightly but statistically significantly higher than in corresponding leukapheresis products (2.33 ± 1.03% vs. 1.84 ± 0.86%, respectively, p = 0.04; Fig. 1A).
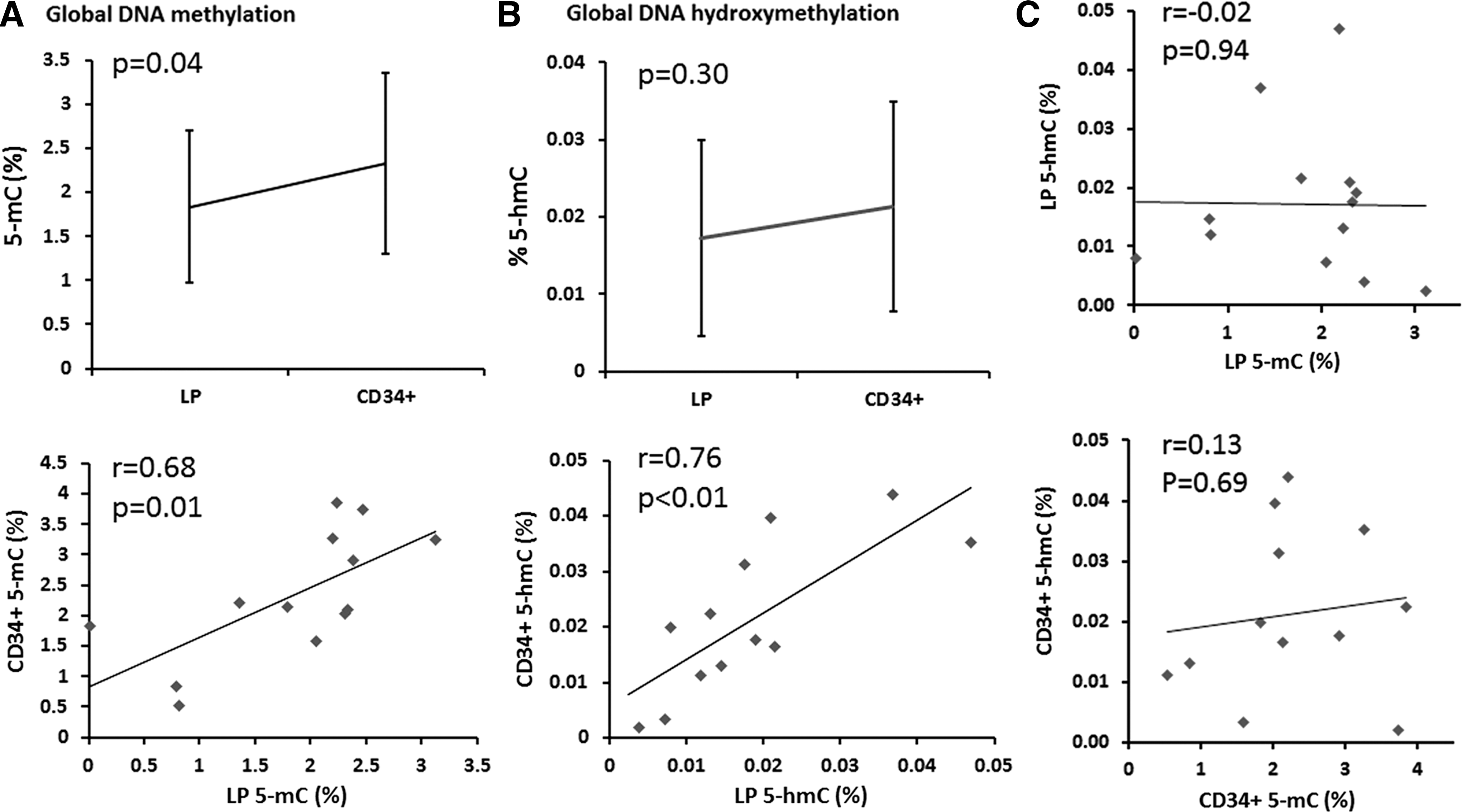
Global DNA methylation and global DNA hydroxymethylation. Global DNA methylation and global DNA hydroxymethylation status was determined by an enzyme-linked immunosorbent assay-based method using MethylFlash Methylated DNA Quantification Kit and MethylFlash Hydroxymethylated DNA Quantification Kit, respectively. Global 5-mC content in CD34+-enriched cell products was statistically significantly higher than in leukapheresis products
Current evidence suggests that high levels of 5-hmC in cellular populations with pluri- and multilineage potency are lost during differentiation (Tahiliani et al., 2009; Ruzov et al., 2011). In adult human tissues, high levels of 5-hmC are mainly restricted to cells of neuronal lineages and bone marrow (Ruzov et al., 2011). As such, we evaluated whether we could detect a significant 5-hmC signal in our bone marrow-derived cells and whether or not the signal is higher in immunoselected CD34+ cells due to their more primitive character. Our results show that 5-hmC is a rare modification in adult cells of hematopoietic origin. The global 5-hmC content did not statistically significantly differ between leukapheresis and CD34+-enriched cell products (0.017 ± 0.013% vs. 0.021 ± 0.014%, respectively, p = 0.30; Fig. 1B).
Both global 5-mC and global 5-hmC content in leukapheresis products correlated well with global 5-mC and global 5-hmC content in corresponding CD34+-enriched cell products (r = 0.68, p = 0.01 and r = 0.76, p < 0.01, respectively; Fig. 1A, B). In line with previous studies (Nestor et al., 2012), global 5-mC content did not correlate with global 5-hmC content either in leukapheresis products or CD34+-enriched cell products (r = −0.02, p = 0.94 and r = 0.13, p = 0.69, respectively; Fig. 1C).
DNA methylation analysis of candidate genes
To select gene pathways and candidate genes for target DNA methylation profiling, we focused on genes, which become demethylated and thereby activated during CD34+ differentiation. To this end, we downloaded the already available high-throughput data from a previously published study (Calvanese et al., 2012), and constructed Venn diagram to identify genes that are marked by DNA methylation loss during CD34+ differentiation (β < 0.7 in CD34-Diff), while being hypermethylated (β ≥ 0.7) in hESCs, CD34-CB cells, and CD34-iPSCs. Out of 13645 genes with β value <0.7 in CD34-Diff, we found 1741 genes, which unlike in hESCs, CD34-CB cells, and CD34-iPSCs, might become activated in CD34+ cells during CD34+ maturation (core set; Fig. 2).
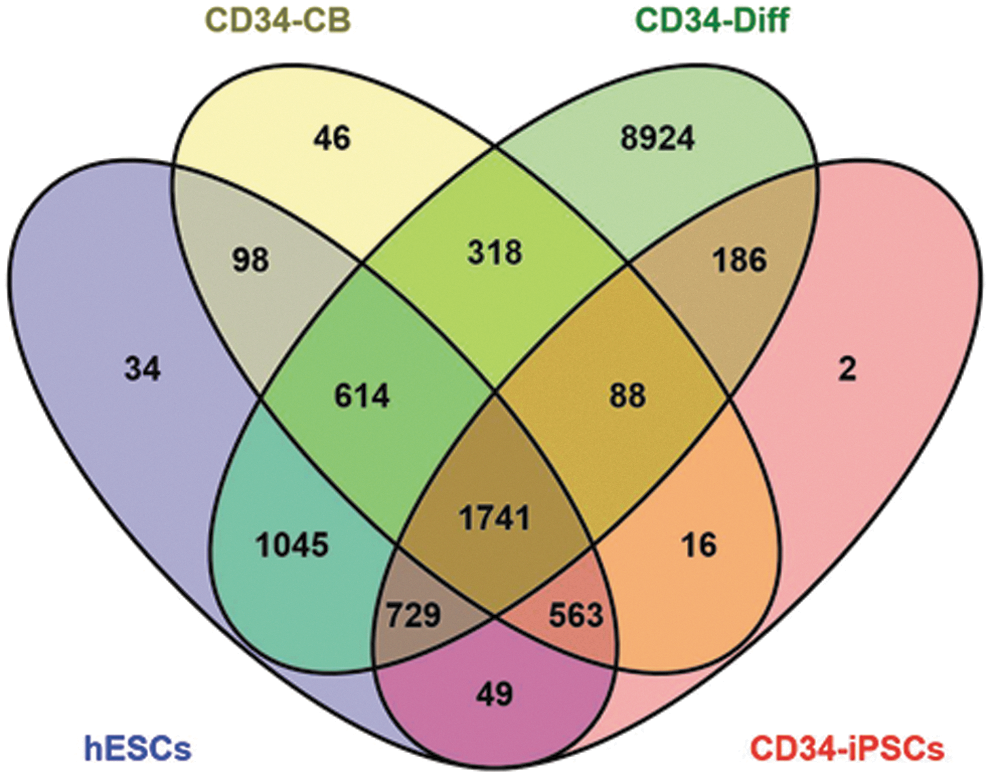
Venn diagram. Four set Venn diagram was constructed with Venny 2.0.2. Computational Genomic Service to identify genes that showed hypomethylation (β < 0.7) in CD34-Diff cells and hypermethylation (β ≥ 0.7) in hESCs, CD34-CB cells, and CD34-iPSCs. Out of 13645 genes with β value <0.7 in CD34-Diff, we found 1741 genes that unlike in hESCs, CD34-CB cells, and CD34-iPSCs might become activated in CD34+ cells during CD34+ maturation (i.e., core set). hESCs—SHEF-1, SHEF-4, SHEF-5, SHEF-7, H7, H14, H14S9, H7S14, HS181, and I3 cells; CD34-CB, CD34+ cells purified from cord blood and pooled from five healthy newborns; CD34-Diff, in vitro differentiated CD34+ cells; CD34-iPSCs, in vitro dedifferentiated CD34+ cells, in which pluripotency was induced by viral-based ectopic expression of the reprogramming factors. hESCs, human embryonic stem cells; iPSCs, induced pluripotent stem cells.
Next, GO analysis was conducted to examine the function of selected CD34+ cell-specific demethylated genes. As expected, GO terms were highly enriched in biological processes such as signal transduction, system process, multicellular organismal development, and anatomical structure development (Table 2; Supplementary Table S1). Due to reports indicating factor-based reprogramming to be less effective at establishing the ground state of pluripotency by displaying DNA methylation signatures specific to their tissue of origin (Kim et al., 2010), we repeated GO analysis disregarding data on hESCs and CD34-iPSCs. Repeated analysis encompassing 318 genes that were hypermethylated in CD34-CB (β ≥ 0.7) and lost methylation marks in CD34-Diff (β < 0.7) again showed nonrandom distribution of GO terms. CD34+ cell-specific demethylated genes were enriched in similar biological terms as previously such as multicellular organismal development, biopolymer metabolic process, anatomical structure development, and signal transduction (data not shown). Guided by the previously described strong general enrichment for potential transcription factor binding sites in all hypomethylated regions (Hodges et al., 2011), we searched for overrepresented transcription factor binding site motifs specific to our core set genes. Pscan software tool identified MAF and NR2F family transcription factors as overrepresented regulators of CD34+ cell-specific demethylated genes (Table 3; Supplementary Table S2). Taken together, our in silico analyses indicate that genes specifically hypomethylated in CD34+ cells are enriched for annotations related to developmental processes.
GO analysis of the core set (i.e., genes that showed hypomethylation (β < 0.7) in CD34-Diff cells and hypermethylation (β ≥ 0.7) in hESCs, CD34-CB cells, and CD34-iPSCs) depicted in Figure 2 using GSEA portal (Broad Institute; Subramanian et al., 2005). Only the top four hits are presented.
FDR, false discovery rate; GO, gene ontology; hESCs, human embryonic stem cells; iPSCs, induced pluripotent stem cells.
As such, we decided to measure promoter DNA methylation of 94 stem cell transcription factors included in the ready-to-use EpiTect Methyl II Complete PCR Arrays. Unsupervised hierarchical clustering of 14 methylation profiles from 8 consecutive samples revealed that methylation profiles of immunoselected CD34+ cells could partly be separated from the methylation profiles of leukocytes in corresponding leukapheresis products (Fig. 3). These findings confirm the presence of a cell- type-specific methylation pattern that shows a very high degree of similarity between individual samples and thus cluster together. Nevertheless, the difference in methylation signatures between leukocytes and immunoselected CD34+ cells was not substantial, and few statistically significant differences appeared (Fig. 3). We identified 15 stem cell transcription factors in which average promoter methylation was statistically significantly different between leukocytes and CD34+ cells (Table 4). The difference in average promoter methylation was highest for HOXC12 (58.18 ± 6.47% vs. 13.34 ± 24.18%, p = 0.0009) and NR2F2 (51.65 ± 25.89% vs. 7.66 ± 21.43%, p = 0.0045) genes.

Heatmap representing promoter DNA methylation of 94 stem cell transcription factors included in the ready-to-use EpiTect Methyl II Complete PCR Array. We used restriction enzyme-based MethylScreen™ technology to profile the methylation status of target genes. Based on the results of the gene ontology analysis, Human Stem Cell Transcription Factors EpiTect Methyl II Complete PCR Array was selected, which profiles the promoter methylation level of a panel of 94 transcription factors whose association with stem cell differentiation and development has been well documented. Unsupervised hierarchical clustering of 14 methylation profiles from 8 consecutive samples revealed that methylation profiles of immunoselected CD34+ cells could partly be separated from the methylation profiles of leukocytes in corresponding leukapheresis products. We identified 15 stem cell transcription factors in which average promoter methylation was statistically significantly different between leukocytes and CD34+ cells. The difference in average promoter methylation was highest for HOXC12 and NR2F2 genes. Hypermethylated genes are represented in red and hypomethylated genes are represented in green.
We used restriction enzyme-based MethylScreen™ technology to profile the methylation status of target genes. Based on the results of the GO analysis, Human Stem Cell Transcription Factors EpiTect Methyl II Complete PCR Array was selected, which profiles the promoter methylation level of a panel of 94 transcription factors whose association with stem cell differentiation and development has been well documented. The table shows the average (plus SD) promoter methylation for 94 transcription factors in leukapheresis products and CD34+-enriched cell products. We identified 15 transcription factors in which average promoter methylation was statistically significantly different between leukocytes and CD34+ cells (shown in gray).
AVG, average; CD34+, CD34+-enriched cell product; LP, leukapheresis product.
NR2F2 gene expression
The current models describing the relationship between DNA methylation and gene expression indicate that promoter methylation is often inversely correlated with gene expression. Methyl marks in promoter region may directly interfere with the binding of certain transcriptional regulators and thereby silence the expression of corresponding genes. Because (1) in silico analysis identified NR2F2 as the second strongest overrepresented transcriptional regulator of CD34+ cell-specific demethylated genes and (2) NR2F2 was among the two transcription factors with the highest difference in promoter methylation between CD34+-enriched cell products and leukapheresis products, we asked whether NR2F2 promoter demethylation increases NR2F2 gene expression in immunoselected CD34+ cells compared to leukocytes. We found that regardless of the NR2F2 methylation status in leukocytes and immunoselected CD34+ cells, NR2F2 promoter demethylation did not result in gene activation/overexpression. In fact, even though a highly sensitive TaqMan method was used for gene expression analyses, there was no NR2F2 expression detectable in any of the samples tested except for positive control (data not shown).
Discussion
Significant changes in DNA methylation pattern have been described during the differentiation processes, in particular when comparing pluripotent stem cells or multipotent progenitors with lineage-committed cells (Mikkelsen et al., 2007). It has been shown, however, that in aging hematopoietic stem cells these epigenetic processes are dysregulated (Chambers et al., 2007), which may lead to functional attenuation of the cells and thereby contribute to impaired outcome of hematopoietic stem cell-based therapies. In our exploratory analysis, we characterized DNA methylation and hydroxymethylation pattern of CD34+-enriched cell products intended for autologous CD34+ cell transplantation in patients with cardiomyopathy. We hypothesized that global DNA methylation and hydroxymethylation profiles differ between more primitive immunoselected CD34+ cells, in CD34+-enriched cell products, and mainly differentiated hematopoietic cells in leukapheresis products (Bocker et al., 2011; Calvanese et al., 2012), and that interindividual differences in the methylation profile of selected genes in CD34+ cells might prove important for cell's regenerative properties and ultimately the clinical outcome of autologous CD34+ cell transplantation.
In accordance with CD34+ cell's more primitive character, we show that global DNA methylation content was slightly higher in CD34+-enriched cell products than leukapheresis products. Unlike reports of high 5-hmC levels present in embryonic tissues as well as brain and bone marrow of adults, global DNA hydroxymethylation content in our immunoselected CD34+ cells was minor and did not differ significantly compared to leukocytes in leukapheresis products. This might imply that 5-hmC levels are high in populations with pluri- and multilineage potency and drop drastically as cells differentiate in a hierarchical manner, or alternatively, our sample size was too low to detect small differences in global DNA hydroxymethylation level between CD34+ cells and leukocytes.
Enrichment for annotations related to developmental processes in genes specifically hypomethylated in CD34+ cells prompted us to analyze promoter DNA methylation pattern of 96 stem cell transcription factors available on a ready-to-use array. This analysis revealed that few differences exist in promoter methylation of stem cell transcription factors between CD34+ cells and leukocytes, suggesting that promoter methylation of stem cell transcription factors can tell us little about the adequacy of CD34+ cells over leukocytes for autologous stem cell transplantation in patients with cardiomyopathy. In addition, no significant difference in promoter methylation of stem cell transcription factors between individual samples supports the view that for a reliable clinical biomarker identification, promoter methylation of other factors or epigenetic characteristics other than promoter methylation should be investigated in aging hematopoietic stem cells. NR2F2, a member of the nuclear hormone receptor superfamily, is, however, worth the additional attention. NR2F2 was identified as the second strongest overrepresented transcriptional regulator in genes specifically hypomethylated in CD34+ cells, and it was among the two transcription factors exhibiting highest difference in promoter methylation level between CD34+ cells and leukocytes. The available published data imply that NR2F2 might regulate the expression of angiopoietin-1 and perturb the angiopoietin-1-TIE2 signaling pathway, resulting in heart and vascular defects observed in NR2F2 mutants (Pereira et al., 1999). This finding is supported by the fact that mice lacking ANG-1 or TIE2 display similar vascular and cardiac phenotypes such as NR2F2 mutants (Suri et al., 1996). Furthermore, NR2F2 is shown to directly regulate angiopoietin-1 expression in cultured cells (Qin et al., 2010). Since angiogenesis has already been proposed by several groups to be responsible, at least in part, for the beneficial effect of autologous CD34+ cell transplantation in patients with cardiomyopathies (Mackie and Losordo, 2011), NR2F2 transcription factor activity in relation to the secretion of angiopoietin-1 by CD34+ cells and clinical outcomes should be investigated in the future.
By linking the observed differences in NR2F2 promoter methylation to changes in gene expression, we found NR2F2 promoter methylation was not associated with NR2F2 gene repression. This finding speaks in favor of a growing body of evidence claiming that active demethylation in human postmitotic hematological cells is not necessarily coupled to alterations in transcriptional activity (Suzuki and Bird, 2008; Klug et al., 2010; Farlik et al., 2016). Alternatively, it is suggested that promoter demethylation keeps the gene in a poised state, that is, not expressed, but maintaining a chromatin state usually associated with the potential for rapid gene activation, for example, in response to external stimuli. A recent study identified increased variability of DNA methylation at loci with properties of promoter and enhancers, especially enriched near genes transitioning between silent and expressed states, and encoding proteins with leukocyte differentiation properties (Wijetunga et al., 2014).
Apart from a relatively small sample size, sampling bias is another disadvantage of our study. Due to strict ethical considerations, we were allowed to collect samples for research purposes of only those patients who mobilized enough cells (more than 80 × 106 CD34+ cells needed for a standard clinical dose). Our study conclusions are therefore limited to good mobilizers. Whether the same findings exist in poor mobilizers is unknown. Second, we cannot exclude the possibility that the G-CSF-induced mobilization had an effect on the methylation pattern or any other aspect of our study.
Conclusion
In summary, this was an exploratory analysis of DNA methylation and hydroxymethylation characteristics in CD34+-enriched cell products intended for autologous CD34+ cell transplantation in patients with cardiomyopathy. We found that global DNA methylation and hydroxymethylation pattern as well as target methylation profile of selected genes in CD34+-enriched cell products does not differ significantly compared to leukapheresis products, and thus can tell us little about the functional capacity and regenerative properties of CD34+ cells. NR2F2 was identified as an interesting candidate to further explore its potential involvement at sites of CD34+ cell engraftment. The ability to understand mechanisms that determine the fate of adult progenitor cells at sites of cell injection may allow us to improve application procedures and clinical outcomes of cell-based therapies.
Footnotes
Acknowledgments
The work was supported by the Slovenian Research Agency (Grant No. J3-5508). We thank Dr. Dragoslav Domanovic for critical comments on the article.
Authors' Contributions
J.Z.C., P.R., and B.V. designed the study; J.Z.C. performed methylation and hydroxymethylation experiments; M.J. and E.M. performed flow cytometry experiments; M.K. collected CD34+ cells for analyses; J.Z.C. collected and analyzed data; M.P.P. and J.Z.C. conducted all statistical and bioinformatics analyses; J.Z.C. wrote the manuscript; P.R. edited the manuscript.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.