
Abstract
8-Oxoguanine DNA glycosylase (OGG1) is responsible for repairing 8-oxo-7,8-dihydroguanine (8-oxoG). Our previous study demonstrated that α-OGG1 protects cells from oxidative damage-induced apoptosis and mitochondrial dysfunction in human lung cancer cells. However, the function of β-OGG1 remains to be elucidated. In this study, we demonstrated that overexpressed β-OGG1 has the same role as α-OGG1 in protecting human bronchial epithelial cells from apoptosis and mitochondrial dysfunction. Furthermore, flow cytometry, confocal microscopy, and western blotting showed that the overexpression of β-OGG1 could block oxidant-induced apoptosis in human bronchial epithelial cells. Additionally, knocking down OGG1 enhanced oxidative damage-induced apoptosis and mitochondrial dysfunction, whereas the overexpression of β-OGG1 had the opposite effects and led to the downregulation of Bax and PARP. The antiapoptotic function of β-OGG1 involved the JNK signaling pathway. These findings suggest that β-OGG1 and α-OGG1 have a similar function on preventing oxidative damage-mediated apoptosis and mitochondrial dysfunction; these effects might be important in the molecular events underlying oxidant-induced cytotoxicity.
Introduction
T
Interestingly, although the levels of the β isoform are nearly 20-fold greater than the α isoform in the mitochondria, it lacks DNA repair activity (Dobson et al., 2000) and its function remains unclear. Nuclear α-OGG1 is highly conserved; its homologs have been characterized in Saccharomyces cerevisiae, Arabidopsis thaliana, Drosophila melanogaster, and some mammals (Klungland et al., 1999; Boiteux and Radicella, 2000). Human α-OGG1 protein has 38% homology with the yeast protein. The β-type OGG1 is only found in human cells (Boiteux and Radicella, 2000). In most human tissues, including the brain, type 1a and 2a are the major OGG1 mRNAs. The C-terminal region of OGG1-2a is essential for proper localization and processing in the mitochondria, and the MTS is essential for its proper targeting to the mitochondria (Nishioka et al., 1999). The important role of OGG1 in mtDNA repair has been highlighted in both in vitro and in vivo studies. Mice deficient in OGG1 (Ogg1–/– ) accumulate 8-oxo-G in both the nucleus and mtDNA, with a 20-fold increase in hepatic mitochondrial 8-oxo-G levels (Cheresh et al., 2015).
mtDNA is subjected to genotoxic assaults from exogenous sources such as exposure to chemotherapeutic drugs as well as from endogenous sources, including the ROS that form as by-products of mitochondrial respiration (Yakes and Van Houten, 1997). The BER pathway is the primary repair pathway in the mitochondrion for restoring DNA integrity (Boesch et al., 2011). Evidence for mitochondrial DNA repair pathways has suggested that mtDNA repair enzymes are encoded by nuclear genes and translocate to the mitochondria (Boesch et al., 2011; Alexeyev et al., 2013). Several chronic human diseases, including diabetes, aging, neurodegenerative disorders (such as Alzheimer's and Parkinson's disease), chronic obstructive pulmonary disease, and cancer, are believed to be associated with mitochondrial dysfunction (Wallace, 2005, 2013; Tuppen et al., 2010; Van Houten et al., 2016; Vyas et al., 2016). Since the mitochondria play a critical role in FeS cluster biogenesis and iron homeostasis, they are likely to be more susceptible to H2O2-induced damage (Yakes and Van Houten, 1997).
Our recent study demonstrated that OGG1 is necessary for maintaining lung alveolar epithelial cell (AEC) mtDNA integrity that is crucial for preventing intrinsic apoptosis following asbestos- or H2O2-induced oxidative stress (Kim et al., 2014). The surprising finding of β-OGG1 being the abundant isoform in mitochondria that lack DNA glycosylase activity led us to hypothesize that OGG1 has a cellular role independent of DNA repair. Our previous study showed that mitochondria-targeted human α-OGG1 (mt-hOgg1) prevented activation of the intrinsic apoptotic pathway in response to oxidative stress not only by augmenting DNA repair but also by preserving mitochondrial aconitase. In addition, the ability of α-OGG1 to prevent oxidant-induced mitochondrial dysfunction and apoptosis was independent of DNA repair (Panduri et al., 2009). We assume that β-OGG1 is important for maintaining mitochondrial genomic integrity. In addition, β-hOGG1 may have a novel role independent of DNA repair in human mitochondria and cells.
The current study demonstrates that β-OGG1 overexpression prevents oxidant-induced mitochondrial dysfunction and apoptosis through a mechanism involving the JNK signaling pathway in human bronchial epithelial cells.
Materials and Methods
Reagents and antibodies
RPMI-1640 culture media, penicillin, streptomycin, and fetal bovine serum (FBS) were purchased from Invitrogen Life Technologies (Carlsbad, CA). For inhibition studies, SP600125 (10 μM) was purchased from Calbiochem (Merck KGaA, Darmstadt, Germany). Antibodies against GAPDH, Bcl-2, PARP, BAX, BAK, p-H2AX, JNK, p-JNK, and Myc were purchased from Cell Signaling Technology, Inc. (Beverly, MA). OGG1 antibody was purchased from Sangon Biotech, Inc. (Shanghai, China). The TUNEL assay kit was purchased from Promega (Madison, WI). pMitoTimer plasmid DNA was purchased from Addgene (
Cell culture
BEAS-2B human bronchial epithelial cells were purchased from the American Type Culture Collection (Manassas, VA). The cells were cultured in RPMI-1640 and supplemented with 10% heat-inactivated FBS, 100 U/mL penicillin G, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
Stable cell line generation
To construct short hairpin RNA (shRNA)-expressing lentiviral vectors for silencing OGG1, the targeting sequence 5′-GCGCAAGTACTTCCAGCTAGA-3′ and stable shRNA expression cassettes were cloned into the PGLV3/H1/GFP+Puro vector, which was purchased from Clontech. Recombinant replication-deficient vesicular stomatitis virus lentiviruses were propagated and purified, and the titer was determined. BEAS-2B cells were then infected with the collected virus supernatant. BEAS-2B cell infections were performed at MOIs of 100 and 200 for 16 h, and the cells infected with PGLV3 (B2B-shcontrol) or PGLV3-OGG1 shRNA (B2B-shOGG1631) were screened using puromycin for 2–5 days. To determine the effect of OGG1 knockdown in shOGG1 cells, western blotting was used to confirm that OGG1 had been knocked down. Lentivirus production and cell infections were performed according to the manufacturer's instructions.
Plasmid construction
The pCMV/Myc/mito plasmids were obtained from Invitrogen. α-OGG1 and β-OGG1 were amplified using c-DNA from normal lung epithelial cells (BEAS-2B) using the following primers: α-OGG1, forward, 5′-GCG
RNA interference
Small interfering RNA targeting OGG1 was synthesized by GenePharma (Shanghai, China). The siRNA sequences were as follows: OGG1 siRNA sense, 5′-GCGGAUCAAGUAUGGACACTT-3′, and antisense, 5′-GUGUCCAUACUUG AUCCGCTT-3′. siRNA transfections were performed using Lipofectamine 2000 reagent according to the manufacturer's protocols.
Cytotoxicity and cell viability assays
Cell viability was assessed using 3-(4,5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2-H-tetrazolium bromide (MTT) assays. A total of 5 × 103 cells/well were seeded in triplicate in 96-well plates. The MTT reagent was prepared at a concentration of 5 mg/mL in phosphate-buffered saline (PBS) and then added to each well at a 1:10 dilution. Cells were incubated for 4 h, and the resulting crystals were dissolved in 100 μL dimethyl sulfoxide. The absorbance at 490 nm was measured using a multiwell plate reader. Cell viability was calculated using the following formula: inhibition ratio = 1 – (OD sample/OD blank control) × 100%.
Measuring apoptosis using flow cytometry
To analyze cell apoptosis, BEAS-2B cells (transfected with negative control (NC) siRNA, si-OGG1, and OGG1 vector) were seeded in six-well plates at a density of 1.5 × 105 cells/well, grown for 24 h, incubated with 100 μM H2O2 for 24 h to induce apoptosis, and then harvested and washed with cold PBS. The cell surface phosphatidylserine in apoptotic cells was quantified using an Annexin V-APC/7-AAD Double Staining Apoptosis Detection Kit (Lianke Bio, Hangzhou, China) according to the manufacturer's instructions. The percentage of apoptotic cells was analyzed using flow cytometry. Triplicate experiments with triplicate samples were performed.
Mitochondrial membrane potential assays
The mitochondrial membrane potential (MMP) (ΔΨm) was measured using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazole-carbocyanide iodine staining (JC-1; Beyotime, Shanghai, China). BEAS-2B cells were transfected with plasmid or siRNA with treatment with H2O2 and incubated with an equal volume of 5 mg/L JC-1 staining solution for 20 min at 37°C. The fluorescence intensity of cells was then analyzed using flow cytometry and the images were viewed and scanned using a confocal laser microscope (TCS-SP5, Leica, Germany) with excitation at 514 nm and emission at 529 nm for green fluorescence and 585 and 590 nm, respectively, for red fluorescence. Red emission represented potential-dependent aggregation in the mitochondria, which reflected ΔΨm. Green fluorescence, appearing in the cytosol after mitochondrial membrane depolarization, represented the monomeric form of JC-1. The ΔΨm of cells in each treatment group was calculated as the ratio of red to green fluorescence.
Determination of mitochondrial DNA copy number
Genomic DNA was extracted using the TaKaRa MiniBEST Universal Genomic DNA Extraction Kit 5.0 according to the manufacturer's protocol (TaKaRa, Japan). The ND1 (mitochondria) and β-globin (nucleus) content was quantified using quantitative polymerase chain reaction (qPCR). The relative expression of ND1 to β-globin was used to represent the mitochondrial DNA copy number (mtDNAcn). Data were analyzed using the 2–ΔΔCt method.
pMitoTimer immunofluorescence
A plasmid containing the pMitoTimer reporter gene was purchased from Invitrogen. The pMitoTimer reporter gene was engineered by targeting a fluorescent Timer protein to the mitochondria by adding the MTS of the cytochrome c oxidase subunit VIII gene to the N terminus of the coding region of Timer, all under the control of a constitutive cytomegalovirus (CMV) promoter. Timer encodes a DsRed mutant (DsRed1-E5) that fluoresces like green fluorescence protein when newly synthesized and shifts the fluorescence spectrum irreversibly to red (Terskikh et al., 2000). The cells were incubated with a mixture of 1 μg of plasmid DNA and 3 μL of Lipofectamine 2000 in Opti-MEM media for 5 h at 37°C; the media were then changed to normal growth medium and incubated overnight. The cells were washed twice with PBS, fixed with 4% paraformaldehyde for 15 min on ice, and then washed twice for 5 min each with ice-cold PBS. The coverslips were mounted on glass slides and images were acquired using confocal microscopy (Leica) with both green (excitation/emission 488/518 nm) and red (excitation/emission 543/572 nm) channels.
TUNEL assays
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was used to analyze DNA damage with the DeadEnd Colorimetric TUNEL System (Promega) according to the manufacturer's protocol. After transfection and treatment, BEAS-2B cells on glass slides were briefly fixed and permeabilized. After equilibration, the cells were incubated in rTdT reaction mixture for 60 min at 37°C. Finally, the slides were stained with 4,6 diamidino-2-phenylindole and mounted with antifade solution, coverslip, and nail polish. After drying the slides, confocal imaging was performed using a confocal laser microscope (Leica).
Western blotting
Cells were harvested and resuspended in an Sodium dodecyl sulfate (SDS) buffer (Beyotime) to prepare total protein extracts. Western blotting was performed according to the manufacturer's protocol (Cell Signaling Technology).
Statistical analysis
The results are presented as mean ± standard error of the mean. Differences between means were analyzed using two-tailed Student's t-tests and were considered statistically significant when p < 0.05.
Results
Effect of down- and upregulating mitochondrial β-OGG1 expression on BEAS-2B cell viability
The reduction in OGG1 protein expression caused by OGG1 shRNA transfection in BEAS-2B cells (also called shOGG1631 stable BEAS-2B cell lines) was confirmed by western blotting. Transfection with OGG1 shRNA reduced OGG1 expression by 30% in total cell extracts and by 80% in mitochondrial extracts compared with vector control-transfected cells (Fig. 1A, B). Next, BEAS-2B cells were transfected with mitochondrial-targeted α-OGG1, β-OGG1, or empty vector. Figure 1C shows the construction of the two isoforms of mitochondrially targeted OGG1. To demonstrate that OGG1 was specifically expressed in the mitochondria, mitochondrial extracts were isolated from transfected cells and then analyzed using western blotting. Compared with vector-only-transfected BEAS cells, mitochondrial extracts overexpressed α-OGG1 and β-OGG1 by >100-fold, as determined by performing western blotting with anti-Myc antibodies to detect α-OGG1 and β-OGG1 in mitochondrial fractions (Fig. 1C, D).
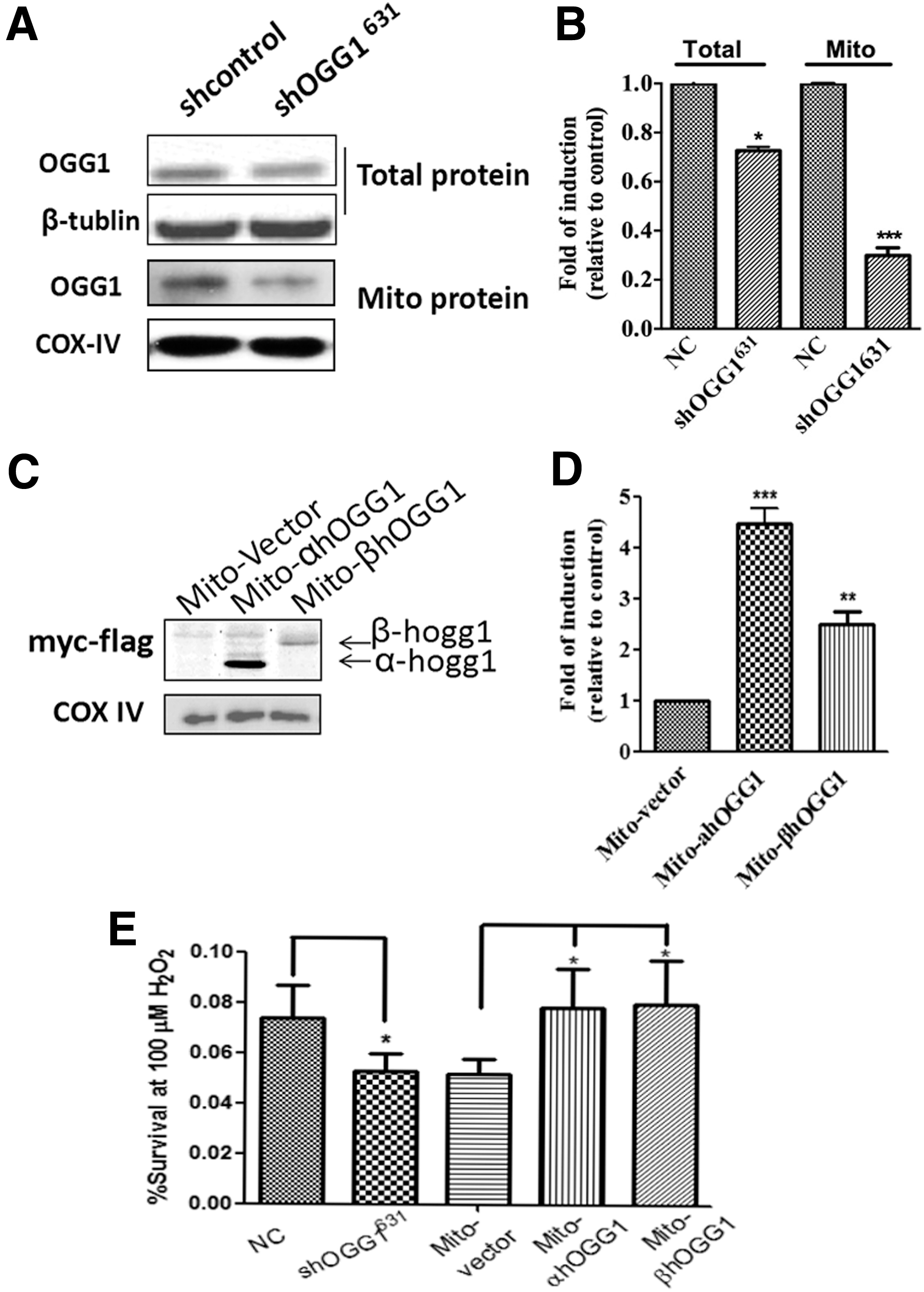
Effects of down- and upregulating mitochondrial β-OGG1 on BEAS-2B cell viability.
Mitochondrial DNA damage can alter mitochondrial function and consequently affect cell growth. Changes in mitochondrial function and cell viability can be assessed using MTT assays. Next, we determined whether downregulating or overexpressing ogg1 in mitochondria affected cell survival following oxidative stress (100 μM H2O2). shOGG1631 cells were more sensitive to oxidative damage than shControl cells. In addition, BEAS-2B cells transfected with α-OGG1 or β-ogg1 were significantly less sensitive to the oxidative damage induced by 100 μM H2O2 than those transfected with mito vector (Fig. 1E); Student's t-tests revealed that the p values for shOGG1631(vs. control), α-OGG1, and β-OGG1 (vs. mito vector) were 0.0004, 0.008, and 0.029, respectively.
Reduced mitochondrial β-OGG1 expression promotes apoptosis
The expression of apoptosis-related proteins was measured using western blotting after treatment with 100 μM H2O2 for 24 h. shOGG1631 increased the expression of Bax and Bak. However, Bcl-2 expression was decreased (Fig. 2A). In contrast, cells transfected with β-OGG1 exhibited reduced Bax and Bak expression compared with those transfected with mito vector or α-OGG1. However, Bcl-2 expression was increased in cells transfected with either α-OGG1 or β-OGG1 (Fig. 2B). To examine the effect of OGG1 on oxidative stress-induced cell apoptosis, apoptosis assays were performed using the Annexin V-APC/7-AAD staining method. The results demonstrated that the downregulation of OGG1 significantly accelerated H2O2-induced cell apoptosis compared with NC (the NC group) in shOGG1631 cells (Fig. 2D, E).
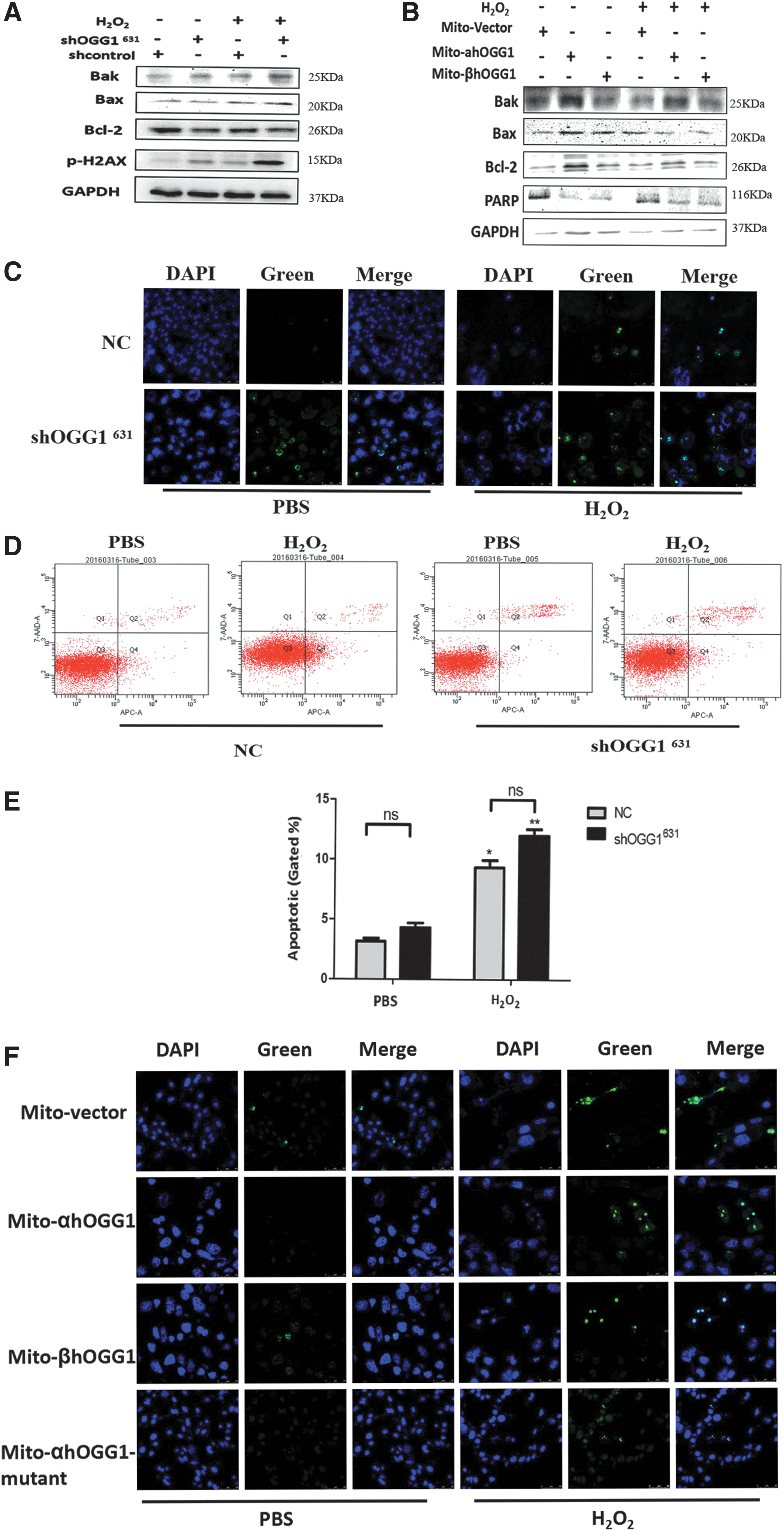
Reduced mitochondrial β-OGG1 expression promotes apoptosis and DNA damage. Control and shOGG1631 cells or mito-α/β/α-mutant-OGG1-transformed cells were incubated with 100 μm H2O2 for 24 h. Apoptosis and DNA damage were assessed using western blotting
β-OGG1 downregulation increases oxidative stress-induced DNA damage
The effect of β-OGG1 on DNA repair protein abundance of H2O2-treated cells was studied to determine if the downregulation and overexpression of OGG1 in the mitochondria affect DNA repair after H2O2 treatment. Levels of the DNA damage marker p-H2AX were increased in shOGG1631 cells treated with 100 μM H2O2 for 24 h. PARP levels were decreased in both α-OGG1 and β-OGG1-transfected cells that had been treated with 100 μM H2O2 for 24 h (Fig. 2A, B). TUNEL assays showed that more shOGG1631 cells had DNA damage compared with those in the NC group (Fig. 2C). When α-OGG1 and β-OGG1 transfectants were treated with H2O2, there were fewer positive cells than with PBS treatment. However, there was no significant difference between mito-mutant OGG1 and vector-transfected cells (Fig. 2F).
Effects of β-OGG1 on oxidative stress-induced mitochondrial damage
Next, quantitative real-time PCR was used to analyze mtDNA integrity in cells in which OGG1 had been knocked down or mitochondrial-targeted OGG1 had been overexpressed and then exposed to oxidative damaging agents. A reduced mtDNAcn is associated with mitochondrial dysfunction, which is a common indicator of apoptotic signaling and can lead to loss of viability (Miller et al., 2003). The results showed that the mtDNAcn was significantly decreased in shOGG1631 cells and increased in the overexpressing β-OGG1 group after treatment with H2O2 (Fig. 3A). Then, quantitative real-time PCR was used to amplify the nuclear β-actin gene and specific mtDNA regions (cytochrome c oxidase [COX-I and COX-II]) as internal controls using DNA isolated from mito vector, α-OGG1 and β-OGG1ls-transfected cells, and shOGG1631 cells treated with 100 μM H2O2 for 24 h. The extent of the decrease in mtDNA integrity was analyzed by calculating the mtDNA/nuclear DNA (β-actin) ratio and then normalizing to untreated controls in which the ratio was set at 100%. The mtDNA/nDNA ratio was used to obtain the relative DNA integrity; a lower ratio represents low initial template and denotes a decrease in the integrity of the mtDNA. Figure 3B shows that the COX-I/β-actin and COX-II/β-actin ratios were significantly decreased in shOGG1631 cells and increased in the overexpressing α-OGG1 and β-OGG1 group compared with their respective control groups.
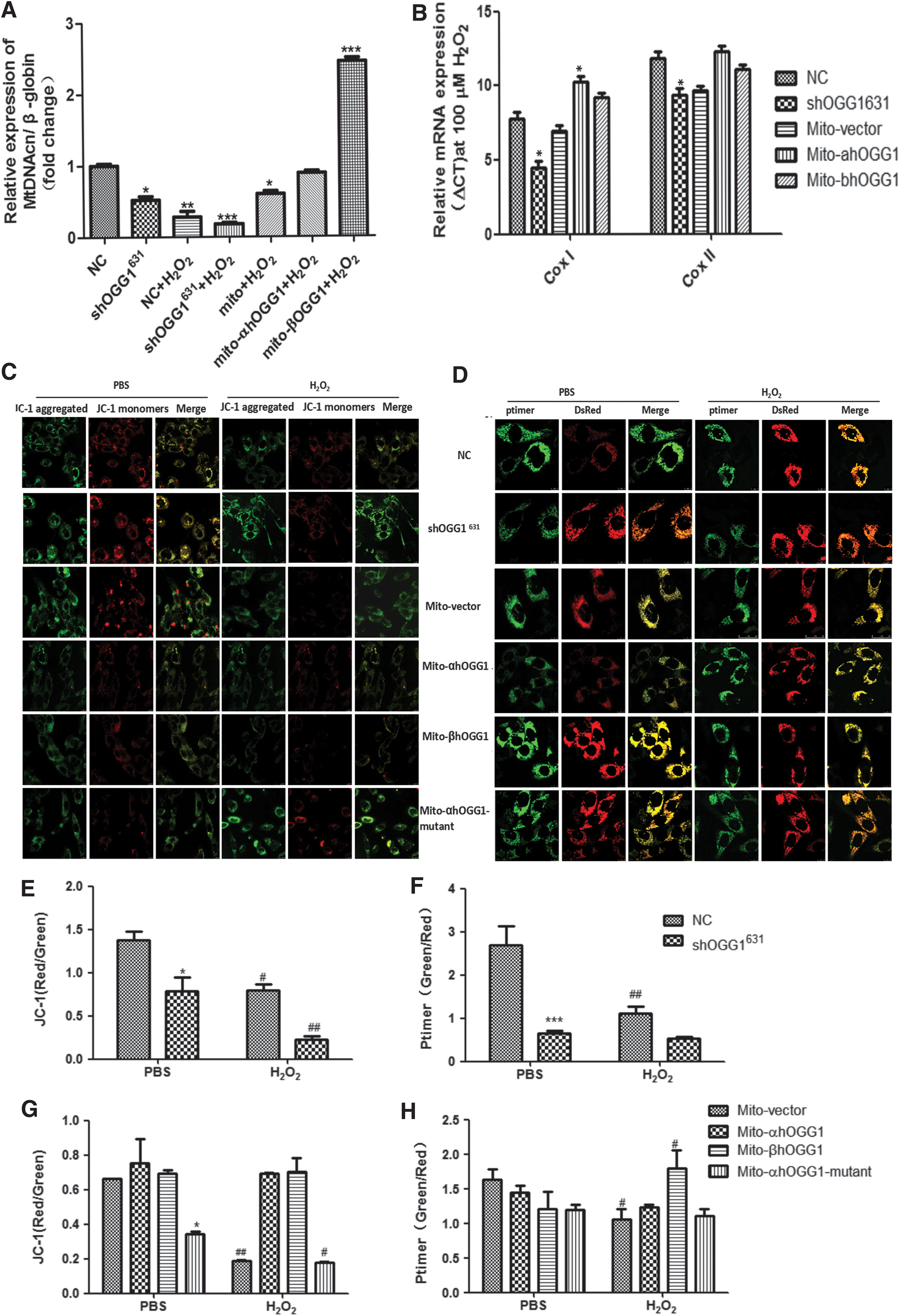
Effects of β-OGG1 on oxidative stress-induced mitochondrial damage. Control and shOGG1631 cells or mito-α/β/α-mutant-OGG1-transformed cells were incubated with 100 μm H2O2 for 24 h.
To determine whether β-OGG1 reduces the overexpressing MMP decline that is caused by H2O2, the fluorescent dye JC-1 was used to measure MMP following H2O2 treatment (Fig. 3C, E, and G). The green/red fluorescence ratio represents a decline in MMP. The results demonstrated that MMP was significantly reduced in shOGG1631 cells compared with the control group (Fig. 3E) and mito-mutant-OGG1-transfected cells compared with the mito vector-transfected group (Fig. 3G). Furthermore, MMP H2O2 exposure decreased the MMP compared with the NC group. However, there was no significant difference in MMP between α-OGG1- and β-OGG1-transfected cells (Fig. 3G).
The shift in the fluorescence spectrum can be calculated by analyzing the ratio of red to green fluorescence in each positive pixel cotransfected with the pMitoTimer reporter plasmid. Figure 3D and F shows that treating shOGG1631 cells with H2O2 resulted in a significant (p < 0.05) shift of mitochondrial fluorescence toward the red spectrum. There was no significant difference between α-OGG1- and β-OGG1-transfected cells; however, mito-mutant-OGG1-transfected cells also had a significant (p < 0.05) shift of mitochondrial fluorescence toward red compared with mito vector-transfected cells (Fig. 3H).
Downregulating mitochondrial OGG1 enhances H2O2-induced apoptosis in BEAS-2B cells through the JNK-MAPK signaling pathway
MAPKs play an important role in certain apoptotic signaling pathways. Compared with control untreated cells, H2O2-treated shOGG1631 cells had significantly increased levels of phosphorylated (activated) JNK1/2 protein (Fig. 4A). In addition, pretreating shOGG1631 cells with the JNK inhibitor SP600125 decreased levels of Bax/Bak and PARP and increased levels of Bcl2 (Fig. 4B). When shOGG1631 cells were treated with SP600125 in the presence of H2O2, the green/red fluorescence ratio (which represents an increased MMP) and the ratio of red to green fluorescence intensity with MitoTimer reporter were increased compared with shOGG1631 cells (Fig. 4C, D).
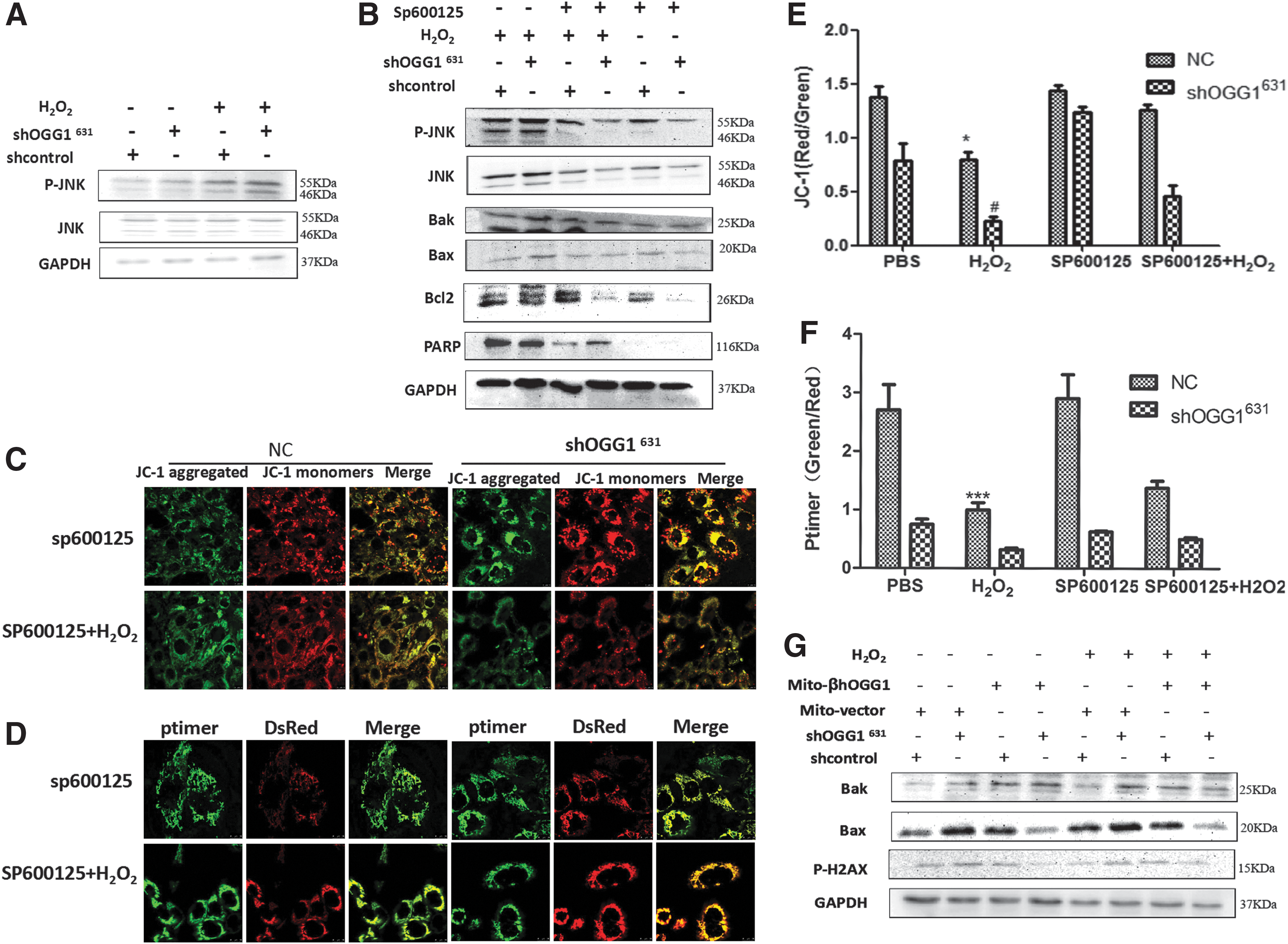
The JNK signaling pathway is involved in OGG1-regulated apoptosis. Control and shOGG1631 stably transfected cells were incubated with 100 μm H2O2 for 24 h in the presence or absence of SP600125 (10 μM).
Mitochondria-targeted β-OGG1 blocks oxidant-induced mitochondrial dysfunction and apoptosis
Finally, western blotting was used to assess apoptosis. shOGG1631 cells transfected with β-OGG1 expressed decreased levels of Bax and the DNA damage marker p-H2AX. These results suggest that β-OGG1 could protect against oxidative stress-induced DNA damage and apoptosis in BEAS-2B cells (Fig. 4E).
Discussion
Previous studies demonstrated that the mitochondrial overexpression of the DNA glycosylase αGG1 suppresses mtDNA damage and cytotoxicity in response to a variety of oxidative stresses in different cells (Dobson et al., 2002; Panduri et al., 2009; Ruchko et al., 2011; Yang et al., 2015). The current study investigated the function of β-OGG1 using OGG1 knockdown stable cell lines and the overexpression of β-OGG1. Its effects on nuclear and mitochondrial DNA damage and cell death in response to the ROS generated by H2O2 and the signaling pathways involved were investigated.
The major mitochondrial OGG1 isoform, OGG1-2a (also named β-OGG1), has been purified; however, its function is unclear. Su et al. (2013) suggested that β-OGG1 was an accessory factor in mitochondrial complex I and was related to mitochondrial BER. To test the function of β-OGG1, stable OGG1 shRNA BEAS-2B cell lines (termed shOGG1631and shcontrol cells) were developed. Since β-OGG1 is more than 20-fold than α-hOGG1 in mitochondria, it suggests that mitochondrial β-OGG1 may be partially knocked out. In the current study, we analyzed the function of β-OGG1 by overexpressing α-OGG1 and β-OGG1 in mitochondria (termed mito-α-OGG1 and β-OGG1).
An important observation in the current study is that overexpressing β-OGG1 lacking a key amino acid in the C-terminal α-O helix that is necessary for 8-oxo-7,8-dihydroguanine (8-oxoG) DNA repair prevented oxidant-induced apoptosis, increased survival, and decreased DNA fragmentation in a manner comparable with α-OGG1. Our previous study showed that mitochondrial α-OGG1 functions have a DNA repair independent in response to oxidative stress by no DNA repair pathway (Panduri et al., 2009). Similar to α-OGG1, β-OGG1 protected cells from oxidative damage-induced apoptosis. Hashiguchi et al. (2004) purified recombinant β-OGG1 and reported that it did not exhibit any significant OGG1 activity in vitro. In another study, Roldan-Arjona et al. (1997) purified recombinant β-OGG1 and showed that its OGG1 activity against 8-oxoG was very low, further indicating that the protective effects of β-OGG1 occur independent of DNA repair.
After H2O2 treatment, the survival rate of shOGG1631 cells was lower than the control group. However, the overexpression of α-OGG1 and β-OGG1 could increase cell survival compared with the vector-only overexpressing group (Fig. 1E). This suggests that β-OGG1 protects against oxidative damage-induced apoptosis; the protective role of α-OGG1 against apoptosis has been reported previously (Tuppen et al., 2010; Alexeyev et al., 2013). Our previous study confirmed that α-OGG1 protects mitochondria in A549 cells (Panduri et al., 2009). The current study assessed the effects of β-OGG1 on mitochondrial function and found that the overexpression of β-OGG1 prevented mitochondrial damage and dysfunction in human bronchial epithelial cells. However, we were unable to successfully knockdown β-OGG1 using shRNA in BEAS-2B cells because of the challenges associated with distinguishing between α-OGG1 and β-OGG1 using short shRNA. Therefore, shOGG1631 silences total OGG1 and does not specifically target β-OGG1.
mtDNA is one of the cellular components that is most affected by oxidative stress. A decrease in mtDNAcn is closely associated with many diseases (Van Houten et al., 2016; Vyas et al., 2016) and has a close relationship with cancer diagnosis and prognosis (Wallace, 2005, 2013). mtOGG1 is also linked to the maintenance of cellular respiratory function and the growth of hepatoma cells (Lee et al., 2013). Compared with nuclear DNA, mtDNA is far more susceptible to ROS-mediated damage because of its proximity to the electron transport chain and the relatively limited mitochondrial DNA repair capacity (Bohr, 2002). The overexpression of mitochondria-targeted Ogg1 prevents mtDNA damage and intrinsic apoptosis in response to a variety of oxidative stresses in various cell types (Dobson et al., 2002; Santos et al., 2003; Ruchko et al., 2011). However, it was unclear whether β-OGG1 modulates the mtDNA damage that affects intrinsic apoptosis. The current study showed that β-OGG1 can inhibit the decrease in mtDNAcn induced by H2O2.
Accumulating evidence has identified a direct association between mtDNA damage and mitochondria-regulated (intrinsic) apoptosis. Notably, H2O2 exposure failed to damage mtDNA in cells transfected with α-OGG1 or β-OGG1, as revealed by qPCR and immunofluorescence staining. This suggests that β-OGG1 and α-OGG1 have the same effects against oxidative stress-induced DNA damage. Mitochondrially targeted OGG1 protects against ROS-induced mtDNA damage and cell death, which suggests that the initial glycosylase-mediated step in BER could be rate limiting in the cytotoxic response to oxidative stress.
A change in MMP is the earliest change in mitochondrial function. A decline in MMP can lead to mitochondrial dysfunction, followed by a loss of viability; it is a common indicator of apoptotic signaling (Yakes and Van Houten, 1997). The current study revealed that the MMP was significantly decreased in BEAS-2B cells exposed to H2O2 and that the overexpression of β-OGG1 could reduce this decrease in MMP; therefore, knocking down the BER enzyme OGG1 can increase MMP. This suggests that β-OGG1 is necessary for maintaining MMP stability. Consistent with our previous report linking α-OGG1 to A549 cell damage (Panduri et al., 2009), this could be a more complete mitochondrial protection mechanism. These functions of β-OGG1 also explains that β-OGG1, which lacks 8-oxoG DNA glycosylase activity and is more than 20-fold than α-hOGG1 in mitochondria, suggests that mitochondrial β-OGG1 may also maintain mitochondrial integrated. A previous study confirmed that OGG1 knockdown augmented mtDNA lesions and intrinsic apoptosis at baseline in AECs and that these effects were enhanced after exposure to oxidative stress (Kim et al., 2014).
The current study revealed that the levels of Bax and Bak were upregulated, Bcl-2 was downregulated, and apoptosis was significantly increased in H2O2-treated shOGG1631 BEAS-2B cells. In addition, the levels of phosphorylated H2AX were increased. These results are consistent with previous reports showing that low-intensity H2O2 treatment induced apoptosis in A549 cells by modulating Bcl-2, Bax, and caspase-3 expression (Panduri et al., 2009). The phosphorylation of histone H2AX on serine 139 (γ-H2AX) is a hallmark of DNA damage and DNA DSBs (Takahashi et al., 2008). Importantly, the apoptotic γ-H2AX ring colocalizes with broken DNA ends that are labeled by TUNEL, indicating that it is initiated by the early wave of DNA breaks at the nuclear periphery (Solier et al., 2012) and that it plays the same role in oxidative stress (Hashiguchi et al., 2004). Furthermore, the overexpression of β-OGG1 reduced mitochondrial oxidative damage. Our previous study has given several evidences between mitochondrial OGG1 and aconitase function in preserving mitochondrial function and blocking oxidant-induced intrinsic apoptosis (Panduri et al., 2009).
Mitochondrial OGG1 may act as a molecular chaperone for aconitase in a manner similar to frataxin, a mitochondria-localized iron chaperone protein that blocks oxidative stress-induced aconitase iron/sulfur disassembly (Bulteau et al., 2004). Another possible mechanism is that OGG1 blocks the key oxidative modification sites on mitochondrial aconitase that are responsible for activating proteolytic degradation through the mitochondrial Lon protease (Bulteau et al., 2003). Therefore, the next step is to confirm whether β-OGG1 interacts with aconitase. Silencing OGG1 using shRNA increased JNK phosphorylation and augmented apoptosis. It was reported that β-OGG1 is an important factor for oxidative BER in the mitochondria, but not in the nucleus (Panduri et al., 2009). The current study found that β-OGG1 overexpression prevented oxidative stress-induced mitochondrial dysfunction and apoptosis through the JNK pathway.
The JNK signaling pathway has been linked to the DNA damage response and is involved with the post-translational modification of DNA repair proteins (Arnould et al., 2002; Kim and Sharma, 2004). JNK is a member of the MAPK family that has been implicated in ROS- and other stress-induced apoptosis (Verheij et al., 1996; Jang and Javadov, 2014). In the current study, increased JNK activation was observed in H2O2-treated shOGG1631 BEAS-2B cells. Inhibiting JNK confirmed that apoptosis and DNA damage were decreased. Meanwhile, treating β-OGG1-transfected shOGG1631 cells with H2O2 decreased the expression of Bak, Bax, and phosphorylated H2AX. These two experimental results are consistent, suggesting that a lack of β-OGG1 could activate the JNK signaling pathway to induce apoptosis and mitochondrial dysfunction. Although JNK can initiate mitochondrial ROS generation, the direct relationship between β-OGG1 and mitochondrial dysfunction needs to be explored further.
In conclusion, the current results revealed that β-OGG1 overexpression reduced H2O2-induced apoptosis and mitochondrial dysfunction in human bronchial epithelial cells through the JNK signaling pathway. These findings identified a function for β-OGG1 and provide novel insight into the effects of oxidative damage on the BER DNA repair system.
Footnotes
Acknowledgments
This work was supported by the Natural Science Foundation of China (grant no.: 81600049 and NSFC81570062); Guangdong Natural Science Foundation (2016A030313681); Guangdong medical University scientific research fund (grant no.: M2015009), Chinese Government Scholarship (CSC), and Guangdong Medical Scientific Fund (A2017010).
Disclosure Statement
No competing financial interests exist.