
Abstract
Clematis apiifolia, belonging to the Clematis L., is a woody vine native to China. It is characterized as heat resistant and fast growing. To better understand potential mechanisms involved in heat-stress responses in Clematis, we characterized the digital gene expression signatures of C. apiifolia under heat-stress conditions. Using RNA sequencing technology, we sequenced six libraries, three biological replicates of control samples and three of heat-stressed samples. In total, 61,708 unigenes were obtained, 36,447 (59.06%) of which were annotated. There were 1941 differentially expressed genes (DEGs) under heat stress, including 867 upregulated and 1074 downregulated genes. Gene ontology enrichment of DEGs revealed that “metabolic process,” “cellular process,” and “single organism” were the top three functional terms under heat stress. A Kyoto Encyclopedia of Genes and Genomes analysis led to the identification of “protein processing in metabolic pathways,” “phenylpropanoid biosynthesis,” and “biosynthesis of secondary metabolites” as significantly enriched pathways. Among the upregulated genes, heat-shock factors and heat-shock proteins, especially small heat-shock proteins, were particularly abundant under heat stress. The data will aid in elucidating the molecular events underlying heat-stress responses in Clematis L.
Introduction
C
Heat stress is often defined as the rise in temperature beyond a threshold level for a period of time that is sufficient to cause irreversible damage to plant growth and development. In general, a transient elevation in temperature, usually 10–15°C above the ambient temperature, is considered heat shock or heat stress. Plants require physiological and biochemical adaptations to avoid or reduce the damage caused by high-temperature stress and to adapt to changes in the surrounding environment (Wahid et al., 2007). Transcriptome analysis is an effective and widely used technique for exploring genes associated with heat resistance. In switch grass, a transcriptome analysis identified 2000 upregulated and 2809 downregulated unigenes under long-term heat stress (Li et al., 2013). In lotus, a transcriptome analysis found that small heat-shock proteins (sHsps) and genes related to cell morphogenesis were upregulated under heat-shock stress (Liu et al., 2016). High-temperature stress inhibits the synthesis of normal proteins in plants and increases the synthesis of heat-shock proteins (Hsps) (Mittler et al., 2012). Hsps function as molecular chaperones under normal conditions. They help in nascent peptide folding, assembly, transport, and membrane transport under stress conditions. They are also involved in the degradation of misfolded proteins and Hsps repair protein aggregation to restore normal functions under the stress of denatured proteins (Kotak et al., 2007).
Based on their molecular weights, Hsps were divided into five categories: Hsp100, Hsp90, Hsp70, Hsp60, and sHsps (Narberhaus, 2002; Ijaz, 2016). Hsp70 proteins are central components of the cellular network of molecular chaperones and folding catalysts, and they are the most evolutionarily conserved Hsps (Mayer and Bukau, 2005). In plants, sHsps are the most widely distributed. Presently, there are 16 kinds of plant sHsps that have been identified in arabidopsis, rice, poplar, soybean, corn, lily, pea, and plum trees (Sarkar et al., 2009). High-temperature stress can induce the synthesis of sHsps. The heterologous expression of sHsps in plants to improve plant stress resistance is presently achieved through transgenic technology (Wahid et al., 2007). However, it is still important to determine the key sHsps in different species because of their level of diversity.
Heat transcription factors (Hsfs) regulate the expression levels of Hsps genes at the transcriptional level under heat shock and heat stress. The DNA-binding domain and oligomerization domain (HR-A/B) of Hsfs in plants can be divided into three categories: A, B, and C (Takii and Fujimoto, 2016). A wide range of cognitive plant Hsfs was first cloned in tomato (Scharf et al., 1990). HsfA1, which is the main factor in heat-shock responses, can regulate the expression of Hsps and other Hsfs (Mishra et al., 2002). Under heat stress, Hsfs can regulate the expression levels of Hsp101s, Hsp70s, and sHsps in Arabidopsis thaliana (At). Hsf mutants have increased heat sensitivity levels, and plants overexpressing Hsfs have strong heat resistance levels (Li et al., 2005; Charng et al., 2007; Ogawa et al., 2007).
In this study, we used RNA sequencing (RNA-Seq) and quantitative real-time-PCR (qRT-PCR) to identify genes in C. apiifolia seedlings under a 40°C heat treatment. In total, six transcriptomes were used in the analysis and numerous differentially, specifically expressed transcripts of heat-regulated genes were identified. These data provide a valuable foundation for genetic studies on heat stress in Clematis L.
Materials and Methods
Plant materials and treatments
Seedlings of C. apiifolia were collected from the Clematis germplasm nursery at the Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, China. Each seedling was placed in a light incubator for 7 days under 25°C conditions for pre-incubation. Then, they were placed at 40°C (heat stress) and 25°C (control) for 6 h. Three independent biological replicates were included for both heat-stress and control treatments. The seedlings were taken, immediately frozen in liquid nitrogen, and stored at −80°C for subsequent analysis.
RNA extraction and library preparation for transcriptome analysis
Total RNAs were extracted by using Trizol reagent according to manufacturer's instructions (Invitrogen, Carlsbad, CA). The total RNA concentration was quantified by using UV spectrophotometry, and the quality was checked by electrophoresis in a 1% agarose gel. Equal volumes of RNA from each of the three replications of the heat-stress treatment and control were pooled. Six independent paired-end libraries were subjected to RNA-Seq analysis. Paired-end libraries with average insert lengths of ∼200 bp were synthesized by using a Genomic Sample Prep Kit (Illumina, San Diego, CA) according to the manufacturer's instructions. Before cluster generation, library concentration and size were assayed by using an Agilent DNA 1000 Kit (Agilent, Palo Alto, CA) on a 2100 Bioanalyzer. Libraries were sequenced on an Illumina HiSeq 2500 instrument by a customer sequencing service (Novogene, Beijing, China). The raw data are available in the National Center for Biotechnology Information Sequence Read Archive (
Sequencing, de novo assembly, and functional annotation
The raw reads were filtered and cleaned by removing low-quality raw reads and adaptor sequences. Clean reads were assembled into non-redundant transcripts by using Trinity, which has been developed specifically for the de novo assembly of transcriptomes using short reads (Grabherr et al., 2011). Short sequences (<200 bp) were removed to improve quality. The resulting sequences were used in BLAST algorithm-based searches and annotations against the National Center for Biotechnology Information non-redundant protein sequences (NR) (
Identification of differentially expressed genes
HTSeq v0.5.4p3 was employed to count the read numbers mapped to each gene. Reads per kilobase per million mapped reads of individual genes were calculated based on gene lengths and read counts mapped to each gene. The resulting p values were adjusted by using the Benjamini and Hochberg's approach for controlling the false discovery rate. Genes with adjusted p values <0.01 determined by using DESeq were assigned as “differentially expressed.”
GO and KEGG enrichment analysis of differentially expressed genes
A GO enrichment analysis of differentially expressed genes (DEGs) was implemented by using the GOseq R package, correcting for gene length bias (Young et al., 2010). GO terms with corrected p values <0.01 were considered significantly enriched by DEGs. For the KEGG pathways analysis, we used KOBAS software to determine the statistical enrichment of DEGs (Mao et al., 2005; Kanehisa et al., 2008).
Quantitative real-time-polymerase chain reaction
Total RNA extraction was the same as for library construction and was treated with DNase I to avoid DNA contamination. Then, 1 μg of RNA was reverse transcribed by using Superscript II reverse transcriptase (Invitrogen) with an oligo (dT) 15 primer according to the manufacturer's instructions (Tiangen Biotech, Beijing, China). Primers were designed by using Primer premier 5 (
Sequence analysis of Hsp and Hsf genes
A. thaliana Hsp and Hsf genes were downloaded from the TAIR Homepage (
Results
Illumina sequencing and reads assembly
To gain a general overview of the C. apiifolia transcriptome under heat stress, three biological replicates for control (C1, C2, and C3) and heat stress (H1, H2, and H3) treated for 6 h at 40°C were designed for RNA-Seq. A total of 272,065,758 high-quality reads, comprising ∼40.81 Gb of sequence data, were generated from the six cDNA libraries (Table 1). The average Q20 percentage of the six libraries was more than 96.99%, suggesting that the quality of the sequence data was high (Table 1). The G + C percentages of the six libraries ranged from 45.10% (H2) to 45.96% (H3). Information on the clean sequence data is listed in Table 1. Using the Trinity method, the high-quality reads were assembled into 87,838 contigs. The contigs produced 61,708 unigenes, with an average length of 873 bp. The N50 length of the unigenes was 1598 bp (Table 2). The majority of unigenes, accounting for 51.18% of sequence data, ranged from 201 to 500 bp. Of the remaining unigenes, 11,003 (17.83%), 12,357 (20.02%), and 4456 (7.22%) ranged in length from 501 to 1000 bp, from 1001 to 2000 bp, and from 2001 to 3000 bp, respectively, whereas 2309 (3.74%) were longer than 3000 bp (Table 2).
Q20 percentage is proportion of nucleotides with quality value larger than 20; GC percentage is proportion of guanidine and cytosine nucleotides among total nucleotides.
Functional annotation and classification of transcriptome sequences
Multiple databases were used to assign accurate annotation information to the unigenes. A total of 36,447 (59.06%) unigenes were annotated by six databases: NR (35,423, 57.40%) databases, Swiss-Prot (23,995, 38.88%), KEGG (21,282, 34.49%), COG (13,573, 22.00%), and GO (25,579, 41.45%) (Table 3). According to the E value distribution of the top hits in the NR database, 19.88% of the matched sequences showed complete homology ( = 0), whereas 27.99% and 52.14% of the sequences showed strong (between 0 and 1.0e-60) and moderate (between 60 and 1.0e-5) homology, respectively. The similarity levels of 15.72% of the sequences were greater than 80.00%, and 42.21% sequences were between 60.00% and 80.00% (Fig. 1). Based on the NR annotation, 78.81% of the unigenes were annotated with sequences from Vitis vinifera, Prunus persica, Populus trichocarpa, Ricinus communis, Fragaria vesca, Glycine max, and Solanum lycopersicum. Among these, 41.05% unigenes were annotated with sequences from V. vinifera (Fig. 1).
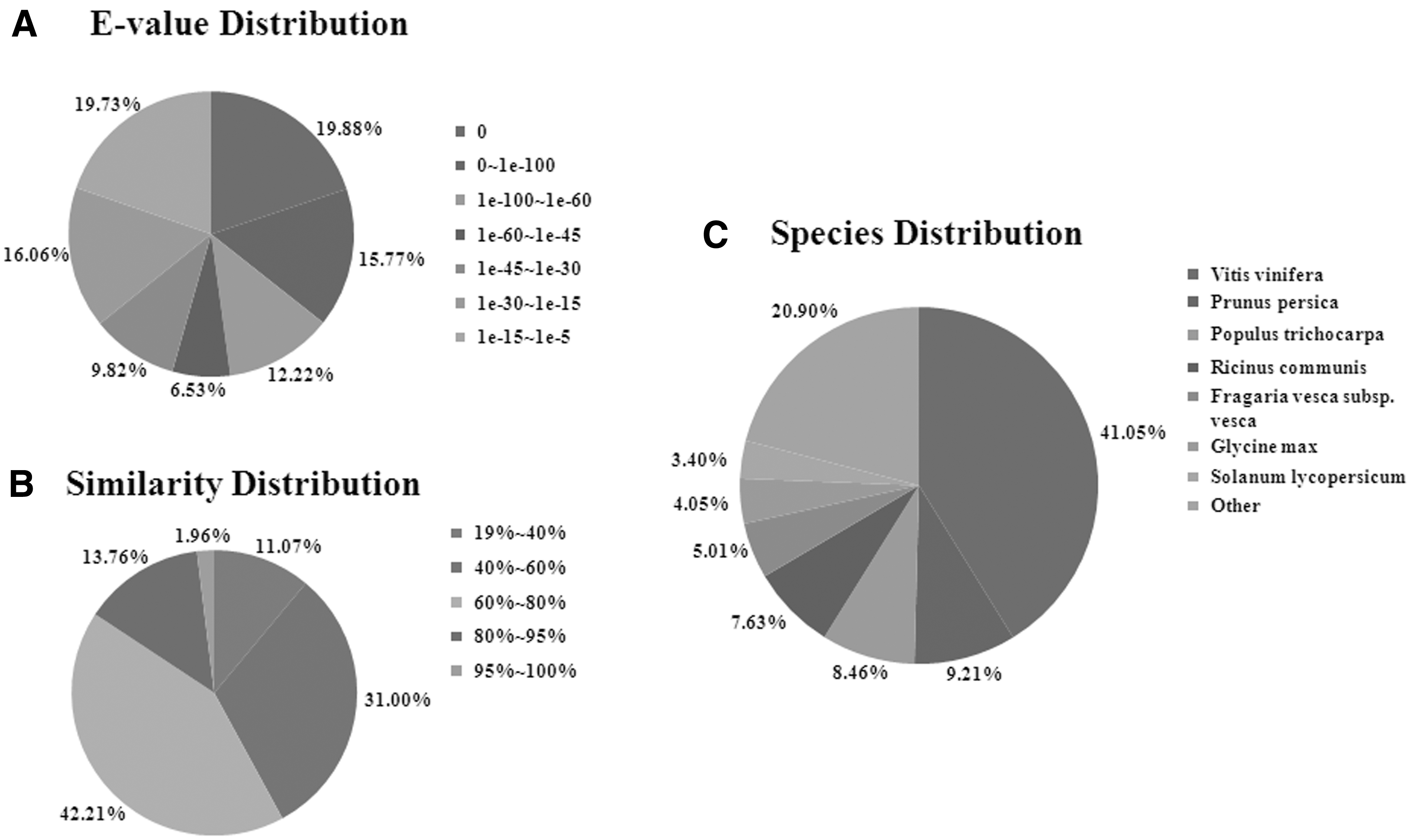
NR classification data.
To functionally classify the genes affected by heat stress, the GO enrichment of the DEGs was investigated by Web Gene Ontology Annotation Plot software. The GO annotated unigenes could be divided into biological process (3132), cellular component (2636), and molecular function (1078) clusters (Fig. 2), and 50 specific terms were significantly enriched (corrected p-value <0.05). Most biology process genes were involved in metabolic processes (594, 18.97%), cellular process (540, 17.24%), and single-organism processes (364, 11.62%) (Fig. 2A). For the cellular component, cell (600, 22.76%), cell part (598, 22.69%), and organelle (453, 17.19%) were the dominant groups (Fig. 2B). Among molecular function, catalytic activity (493, 45.73%) and binding (401, 37.20%) were the dominant groups (Fig. 2C). We mapped the unigenes to the KEGG pathway database and found that 14 pathways (corrected p value ≤0.01) were significantly enriched under heat stress (Fig. 3).
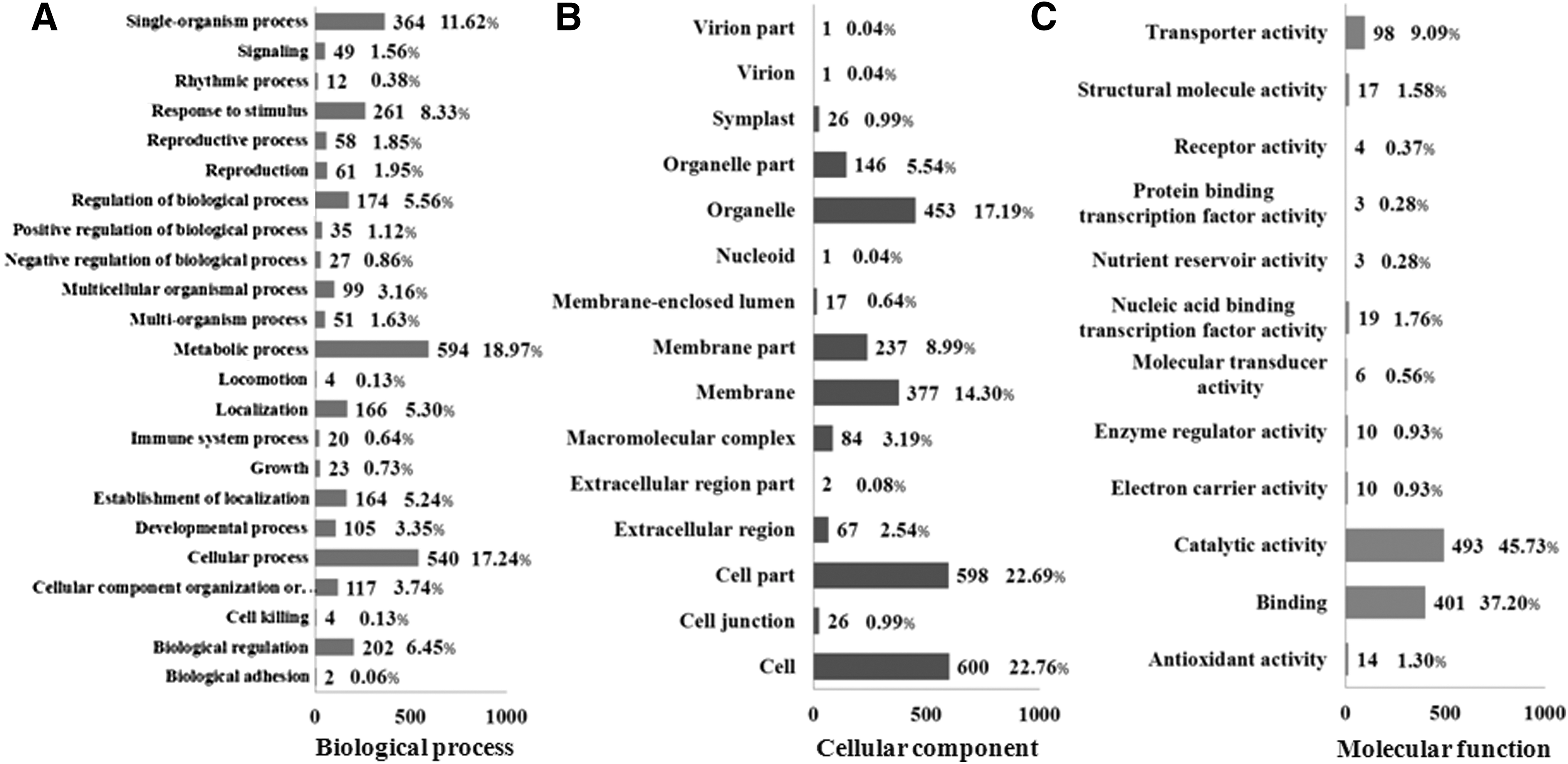
GO classification analysis of unigenes in Clematis apiifolia. GO terms are summarized in three main categories:
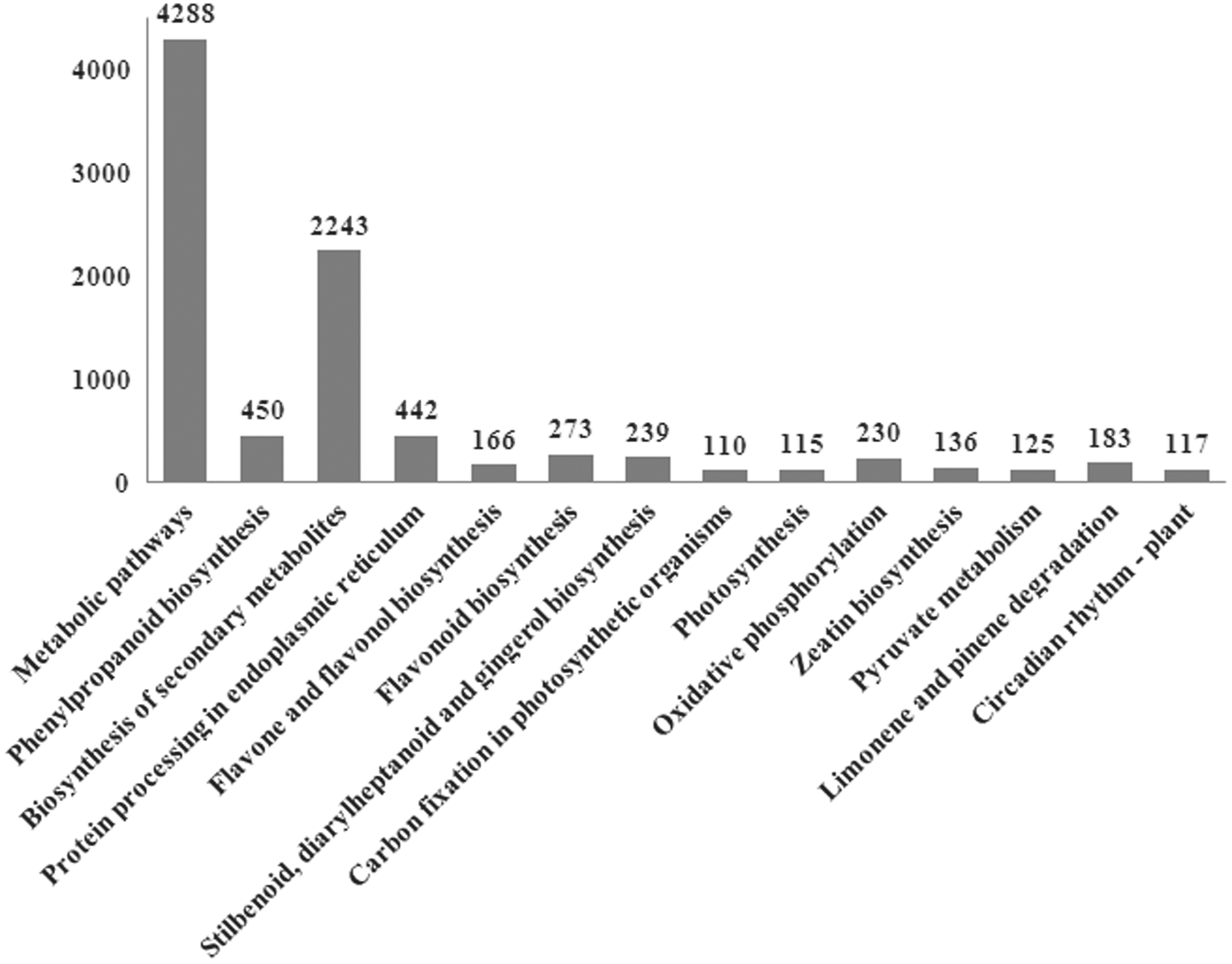
Specific KEGG pathways significantly enriched in DEGs. Pathways with p-values ≤0.01 were considered significantly enriched in DEGs. DEGs, differentially expressed genes; KEGG, Kyoto Encyclopedia of Gene and Genomes.
DEGs and qRT-PCR validation
To identify the DEGs associated with the transition from 25°C to 40°C treatments among our DEGs, we realigned the short reads that were generated from the six independent libraries, which included three libraries under room temperature (25°C) and three libraries under high temperature (40°C), between the two temperature conditions. This process enabled us to evaluate the gene expression abundance levels by calculating the │log2 Ratio│ and to estimate the significance of the DEGs between the two treatments. We observed that no less than 80.00% of the total clean reads in each library could be mapped uniquely to all transcripts. In this study, when the false discovery rate was less than 10–3 and the average │log2 Ratio│ ≥1 between two treatments, then the transcripts were considered statistically significant DEGs. Through a comparative analysis of biological repeats, 1941 DEGs were identified between the treatments. Among the DEGs, 867 were upregulated and 1074 were downregulated (Fig. 4).
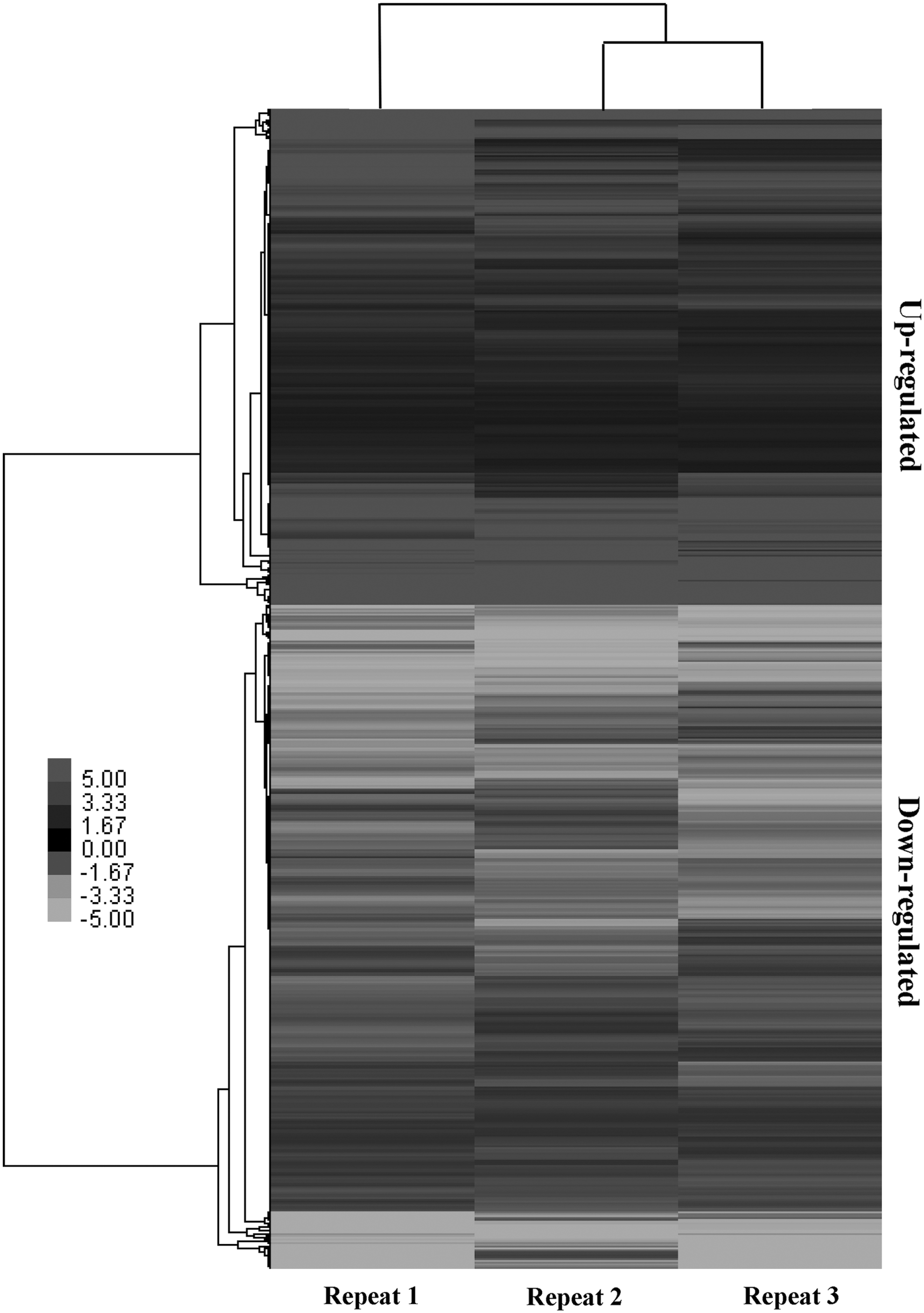
Hierarchical cluster heat map and cluster tree from DEGs. Black indicates upregulated genes, and white indicates downregulated genes.
To validate the DEGs under heat stress, we selected nine significant DEGs for qRT-PCR analysis. The gene expression profiles of these DEGs using qRT-PCR revealed similar variation trends as in the RNA-Seq samples, indicating the high credibility of the transcript abundances of these genes under normal and high temperatures from different individuals (Fig. 5).
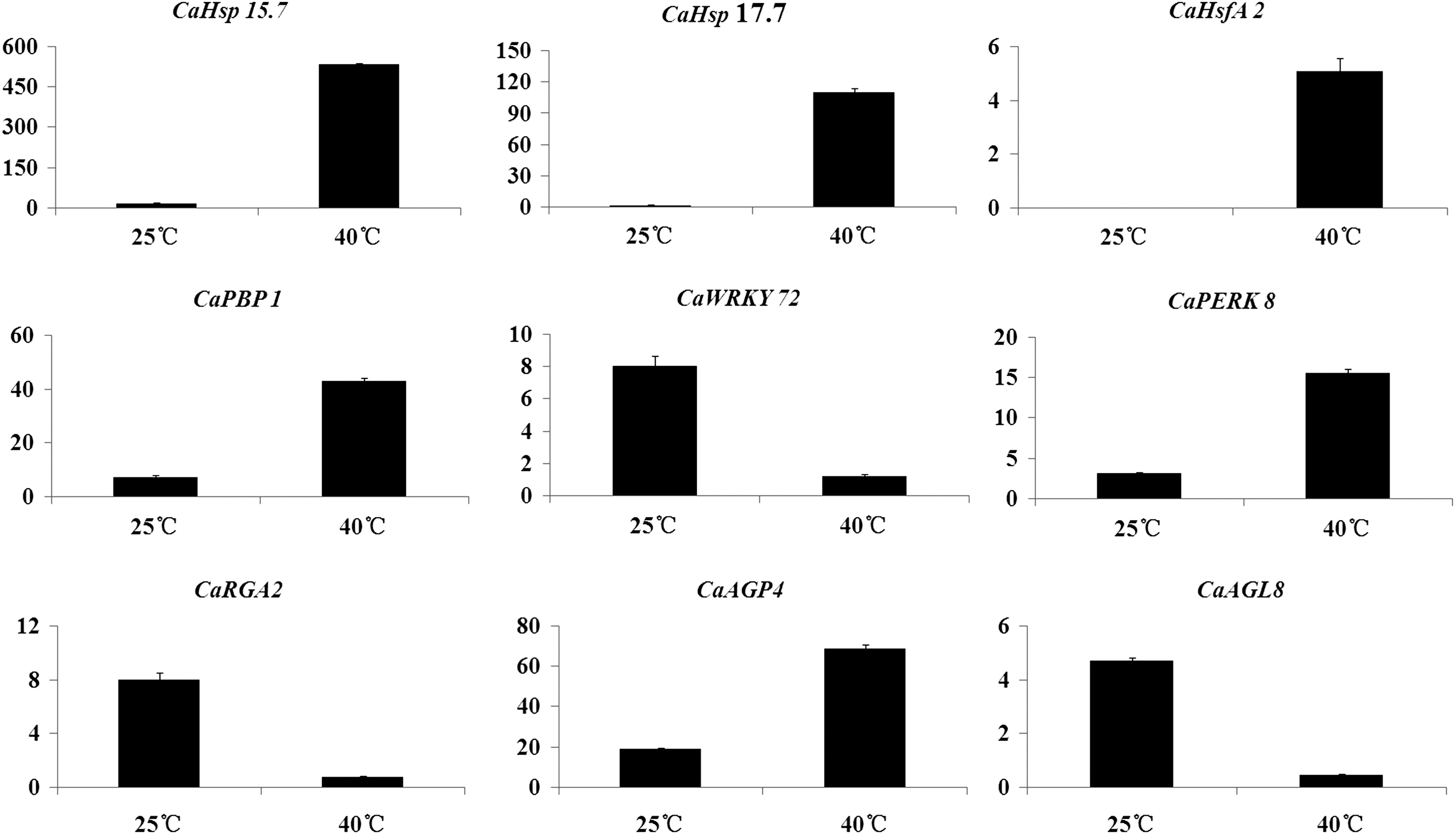
Analysis of genes regulated by heat stress in C. apiifolia using qRT-PCR. Data are the mean ± standard errors (n = 3). qRT, quantitative real time.
Hsps and Hsfs in C. apiifolia
In plants, Hsps play important roles in heat-shock responses. Data revealed the upregulation of Hsps and Hsfs under heat-stress conditions in C. apiifolia. A total of 34 Hsps were identified, including Hsp90, Hsp70, and sHsp family members. Among these, 31 belonged to the sHsp family, accounting for 91.18% of the Hsps identified. In terms of gene expression level, 91.18% were more than threefold increased under heat stress (Table 4). Among 31 sHsp genes, the phylogenetic and conserved motifs of 25 full length genes were analyzed. The 25 CasHsps were classified into eight clades and contained two to eight different conserved motifs (Supplementary Fig. S1). In this study,
Discussion
High temperature is a common abiotic stress for higher plants. RNA-seq is a relatively efficient method for conducting large-scale transcriptomic studies, including gene expression patterns, which are constantly being improved. In this study, to ensure the accuracy of the sequencing data, each treatment had three biological repeats. Six plant pools under heat stress and control conditions were used for the analysis. The base composition and quality analyses showed that the Q20 ratios were above 96.99%, and the G + C contents were above 45.10% (Table 1), which indicated successful library construction and good sequencing quality. Whole clean reads were assembled into 87,838 contigs and 61,708 unigenes of C. apiifolia. The contigs covered 39.71 Mb of transcriptome sequences, with an average length of 452 bp and an N50 length of 1104 bp. The unigenes had a total length of 53,857,292 bp, with an average length of 873 bp and an N50 length of 1598 bp. These findings indicate that the experimental design is reasonable and the sequencing quality is good.
Gene expression profile under heat stress
Responses to heat stress form a complex phenomenon involving extensive gene expression changes in plants. In this study, we focused on the responses of Clematis to long-term stress. The heat-stress conditions of 40°C for 6 h were selected for transcriptomic analysis, and screening for heat shock-related genes was avoided. Overall, 1941 genes were differentially expressed under heat stress in C. apiifolia. After a long-term heat stress of more than 6 h, the proportion of heat-induced genes was different than the 867 genes upregulated and 1074 genes downregulated in our results, as seen in studies of barley, wheat, lotus, and rice (Qin et al., 2008; Mangelsen et al., 2011; Zhang et al., 2012; Liu et al., 2016). In our research, 27.28% of the DEGs remained uncharacterized because no homologs have been identified in the various comparison libraries. These genes had specific responses to heat stress in C. apiifolia.
Roles of Hsps and Hsfs under heat stress
In higher plants, heat stress redirects protein synthesis by decreasing the transcription and translation of normal proteins, and stimulating the synthesis of a new set of proteins: Hsps (Schulze et al., 2005). Hsps are the major functional proteins that are induced by heat stress. The induction of Hsps when plants are exposed to elevated temperatures has been well documented. Although Hsps is important in thermotolerance, the specific functions and targets of Hsps remain largely unknown. Based on their approximate molecular weight, the principal HSPs are grouped into six conserved classes: HSP100/Clp, HSP90/HtpG, HSP70/DnaK, HSP60/GroEl, HSP40/DnaJ, and sHSPs (Schulze et al., 2005). In this study, 34 DEGs of Hsps were grouped into Hsp90, Hsp70, and sHsp family (Table 4). Meanwhile, our results revealed that sHsps were the largest chaperone gene family responsive to heat stress (Table 4).
Hsp21 may interact with the plastid nucleoid protein pTAC5 and is necessary for the development of chloroplasts during heat stress in Arabidopsis (Zhong et al., 2013). Similarly, the unigene homologous with Pisum sativum Hsp21 (Unigene21603) was upregulated under heat stress (Table 4). OsHsp26 plays an important role in the protection of Photosystem II during heat stress (42°C) (Kim et al., 2011). In our transcriptome, the unigene Unigene17345 homologous with G. max Hsp26A was downregulated, indicating that it played a negative regulatory role under heat stress (Table 4). An accumulation of AtHsp17.6A proteins could be detected after heat exposure and at a late stage of seed development, but not under osmotic stress conditions, suggesting the stress-induced post-transcriptional regulation of AtHsp17.6A expression. Interestingly, the unigene homologous with AtHsp17.4b (Unigene16677) was upregulated in our transcriptome, suggesting that it was important for clematis' response to heat stress (Table 4). The overproduction of AtHsp17.6A can increase salt and drought tolerance in Arabidopsis (Sun et al., 2001), and Hsp70 confers tolerance to heat and other abiotic stresses in Arabidopsis (Montero-Barrientos et al., 2010). Mitochondrial Hsp70 may suppress programmed cell death in rice protoplasts by inhibiting the amplification of reactive oxygen species (Qi et al., 2011). In this study, two DEGs belonging to the Hsp70 gene family showed all enhanced mRNA levels after heat treatment (Table 4).
The molecular mechanisms leading to Hsp expression under stresses are not entirely understood, but Hsfs serve as the terminal components of signal transduction mediating the expression of HSPs and other heat stress-induced transcripts are widely accepted (Pockley, 2003; Kotak et al., 2007; Penfield, 2008). HsfA2 was expressed under 6-h heat-stress conditions in our research. The structure and function of HsfA2 are similar to those of HsfA1, but HsfA2 is expressed only in stressed plants. High levels of HsfA2 can be induced during the continuous heat stress and recovery stage in A. thaliana (Nishizawa-Yokoi et al., 2009). The overexpression of AtHsfA2 not only improves the heat resistance but also improves the resistance to oxidative stress and hypoxia caused by salt (Banti et al., 2010). HsfA2 plays an important role in preventing oxidative damage and cell death in plant organelles, which are critical regulators of plant stress responses (Zhang et al., 2009). Here, HsfA2 was upregulated in C. apiifolia, suggesting that it might play a positive role in response to heat stress (Table 5).
Overall, the findings of this study will facilitate a better understanding of the roles of sHsps and Hsfs in the heat-stress response of C. apiifolia.
Conclusion
In this study, we characterized the digital gene expression signatures of C. apiifolia under heat-stress conditions, which led to the identification of significantly enriched pathways and a number of candidate genes involved in heat-stress responses. Based on the data presented here, we hypothesize that the sHsps play a key role in the heat-stress responses of C. apiifolia. Data from this study will aid in understanding the molecular events underlying responses to heat stress in C. apiifolia.
Footnotes
Acknowledgments
This work was supported by the Prospective joint project of Jiangsu Province Science (Grants No. BY2015074-01) and Jiangsu Key Laboratory for the Research and Utilization of Plant Resources (Institute of Botany, Jiangsu Province and Chinese Academy of Sciences) (Grants No. SQ201301).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.