
Abstract
Holt–Oram syndrome (HOS) is an autosomal dominant disorder, which is characterized by deformities of upper limbs and congenital heart defects. Alterations of TBX5 gene have been identified to be the leading cause of HOS, while some cases could not be explained by TBX5 mutations. In our study, we preliminarily diagnosed a newborn baby, who had Tetralogy of Fallot, thumb agenesis, facial dysplasia, and right ear canal malformation, as HOS. Chromosome microarray analyses showed no pathological deletions or replications of chromosome segments; whole exome sequencing screened out six candidate genes that were involved in cardiac diseases or syndromes among which SALL4 has been reported as HOS related gene. We evaluated the pathogenicity of SALL4 mutant sites by series of software. The results indicated that SALL4-M143V may be a polymorphism site, and SALL4-R418C could cause disease. HOPE and SWISS PDB viewer showed that SALL4-R418C leads to changes in amino acid properties, loss of protein hydrogen bond, and functional impact of SALL4 zinc finger domain. These results further confirmed the pathogenic significance of SALL4-R418C mutant. When genetic analyses coupled with bioinformatic analyses, we identified a SALL4 gene rare mutation which might contribute to a newborn with HOS. Although some doubts need to be further discussed and explored, our study deepened the understanding of phenotype difference among syndromes and role of SALL4 mutations in disease occurrence.
Introduction
H
Genetic etiology of HOS has not been fully elucidated, while mutations of genes such as TBX5 and SALL4 are considered to be a relatively credible mechanism of pathogenesis (Aherne et al., 2013; Al-Qattan and Abou Al-Shaar, 2015). TBX5 gene, which located on chromosome 12q24.1 and encodes a protein of 518 amino acids, is a member of the T-box transcription factor family and plays a significant role in embryonic development of heart and forelimb (Steimle and Moskowitz, 2017). TBX5 knockout mice exhibited phenotype of HOS, including malformations of anterior forelimb, septal defects of heart, and dysfunction of cardiac conduction system (Moskowitz et al., 2004). TBX5 mutations have been identified as a crucially genetic basis of HOS; almost 70% HOS patients carry genetic alterations of TBX5 gene (Debeer et al., 2007).
SALL4 is a member of the spalt-like (SALL) gene family (SALL1 to SALL4), which encodes a zinc finger transcription factor and plays an essential role in maintaining the pluripotent and self-renewal properties of embryonic stem cells (Tatetsu et al., 2016). SALL4 could interact with TBX5 to regulate patterning and morphogenesis of embryonic upper limb and heart according to the study of Bruneau (Koshiba-Takeuchi et al., 2006). In some cases, patients who diagnosed with HOS were identified to carry SALL4 mutations, indicating the potential role of SALL4 mutations in occurrence of HOS (Kohlhase et al., 2003). Guo et al. (2015) made a list of genes related to heart–hand defects, including EVC, EVC2, GLI3, HAND2, HOXD13, HRAS, ROR2, SALL1, SPRY4, TBX4, TFAP2B, PAPA2, PAPA3, WNT7A, and ZRS of SHH, mutations of which may also be risk factors of HOS.
In this study, we reported a neonatal HOS patient with the clinical characters of TOF, thumb agenesis, facial dysplasia, and right ear canal malformation. We used chromosomal microarray and whole exome sequencing (WES) technology to detect the genetic alterations of this patient and found a candidate SALL4 mutation. By a series of bioinformatical analyses, we found that the site had a certain pathogenic significance and might take some part of responsibility of the disease.
Materials and Methods
Patients and samples
Since diagnosed with TOF in utero, the proband was performed with a series of examinations after birth, including overall physical examinations, routine blood, urine and stool test, electrocardiogram (ECG), cerebral and ventral doppler ultrasonography, echocardiogram, and chest radiography. Family history of proband was also simply collected.
To further explore the cause, we collected the peripheral venous blood of proband and her parents and extracted DNA using the QIAamp DNA Blood Midi Kit (Qiagen, Dusseldorf, Germany) according to the manufacturer's instruction.
Chromosomal microarray analysis
DNA of the proband was amplified, labeled, and hybridized to the CytoScan HD array platform (Affymetrix, USA) which offered 2,696,550 probes, including 743,304 single nucleotide polymorphisms and 1,953,246 nonpolymorphic probes according to the protocol. Statistic results were analyzed using the Chromosome Analysis Suite software (Affymetrix, USA). The genome version was GRCH37 (hg19).
Whole exome sequencing
DNA of the proband was sent to a commercial provider (Shanghai Biotechnology Co., Ltd., Shanghai, China), which performed sequencing service using the HiSeq 2000 platform. DNA was sonicated into fragments and hybridized to the array. Enriched DNA fragments were eluted and amplified using ligation-mediated PCR. The enriched DNA was then ligated with DNA ligase to fragments ranging in size between 2 and 5 kb randomly; resultant DNA fragments were cut into average 200 bp and were submitted to standard Illumina HiSeq 2000 platform. Image analysis and base calling were carried on with the Illumina's Consensus Assessment of Sequence and Variation 1.8, using the default parameters. According to the quality of reads, the company analyzed the fragments with different tools (Gollob et al., 2002; Li et al., 2009). Final sequencing results were exported in the format of Excel.
Candidate gene and mutation analyses
General information of candidate genes is obtained from GeneCards (
Results
Clinical findings
The proband (G1P1), female newborn with birth weight of 2730 g, was delivered by cesarean section. Echocardiogram showed atrial septum defect and typical TOF signs, including right ventricular hypertrophy, aorta overriding, pulmonary stenosis, and ventricular septum defect. Except from complex malformation of heart, postnatal physical examination also found multiple extracardiac malformations such as thumb agenesis (root of the right thumb and the metacarpal of the proband were connected with pedicle-like soft tissue without joint motion), facial deformity (facial dysplasia, showing distortion of mouse angle when crying, while the resting state was normal), and right auricle and ear canal malformation (Fig. 1). ECG suggested sinus rhythm. Blood tests showed slight electrolyte imbalance. Other examinations, including urine test, stool test, cerebral and ventral doppler ultrasonography, exhibited no special abnormity.
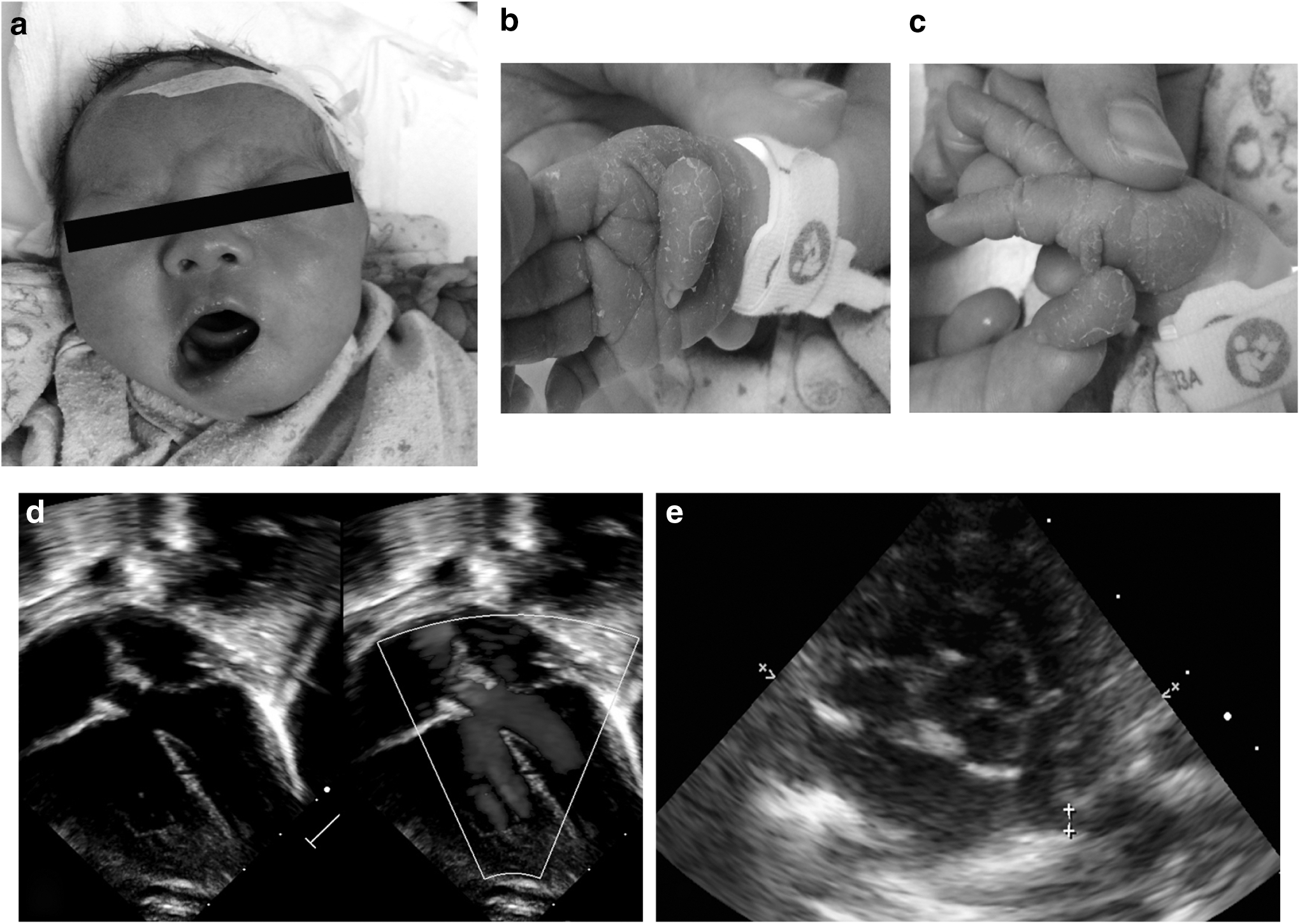
Clinical features of the proband.
The mother of the proband was a 38-year-old woman. She had a threatened abortion during early pregnancy and was treated with tocolytic agents. The parents seemed to be in good health and had no obvious disease characteristics. There was no history of congenital heart disease or syndrome in the family of the proband.
Chromosomal microarray analysis and WES analyses
For CytoScan HD array, quality of the sample data was filtered referring to the following conditions: gain or loss copy number variant (CNV) fragments >50 kb, CNVs contained at least 25 consecutive probes, and finally, compared with known CNVs in the Database of Genomic Variants (
WES experiment exported 92,215 reads (raw data of WES is provided in Supplementary Table S2). We screened the sequencing results according to the following criteria: Quality filter value was “PASS”; mutations located in exonic regions (coding region, 3′ untranslated region, 5′ untranslated region) and gave priority to those in protein coding regions; mutation function excluded synonymous single nucleotide variants (SNVs); and max population frequency was <0.001.
We then used GeneCards combined with PubMed to search for the “identify cards” of screened genes and found two alterations of SALL4 gene which had been identified to contribute to both cardiac and thumb development. The proband carried two SALL4 mutations, SALL4-M143V (c.A427G) and SALL4-R418C (c.C1252T). We first used online software SIFT, PROVEAN, PolyPhen, and MutationTaster to predict the pathogenicity of mutants. We got a consistent result, that is, SALL4-M143V was a tolerant mutant, and another mutation SALL4-R418C was highly possible to cause disease (Table 2).
We also filtrated other five genes, PLXDC1, SELE, TTN, KDM6A, and KIF7, which were related to syndromes or heart development or cardiac diseases, while their relationships with HOS had not been reported. Clinical features of these genes were listed in Table 1. For the mutations of these genes, KIF7 and TTN gene mutations were considered to be pathogenic depending on the online software already mentioned, meanwhile, mutations of PLXDC1, SELE, and KDM6A might be benign (Table 2). We then sequenced the mutant sites of SALL4, KIF7, and TTN genes of the parents to identify the genetic significance of these mutations, and the results are showed in Table 3.
/, not involved.
/, not involved; AA, amino acid alterations of mutations; B, benign; CDS, coding sequence alterations of mutations; D, damaging; MAF, minor allele frequency.
Biological and functional analysis of SALL4 mutations
Considering gene function and pathogenic significance of its mutation, we thought that SALL4-R418C was comparatively more likely to be associated with disease. We then further analyzed the biological and functional significance of SALL4-R418C. By listing amino acid of targeted site in different species in MutationTaster, we found that the site involved in our study of SALL4 was highly conserved among species, including vertebrates and invertebrates, indicating a probably biological function of this site (Fig. 2a). HOPE software indicated that amino acid properties were altered by mutant SALL4-R418C. The mutant residue was smaller than the wild-type residue. The wild-type residue charge was positive; the mutant residue charge was neutral. The wild-type residue was more hydrophobic than the mutant residue (Fig. 2b, c). Alteration of these properties might cause loss of interactions with other molecules of wild-type protein or affect hydrogen bound formation. HOPE also indicated that the mutated residue was located in a Zinc-finger domain that was important for binding of other molecules. The mutation might disturb the interaction and affect the function of the protein. Using SWISS PDB viewer, we constructed the spatial conformation of SALL4 wild-type protein and its mutant SALL4-R418C. We found that wild-type Arginine in position 418 was connected to glycine in position 406 with a hydrogen bond, while mutant cysteine was not in the correct position to make the same hydrogen bond as the original wild-type residue did (Fig. 2d, e). Such bond reduction might influence the stability of protein or influence the binding ability of SALL4 Zinc-finger domain.
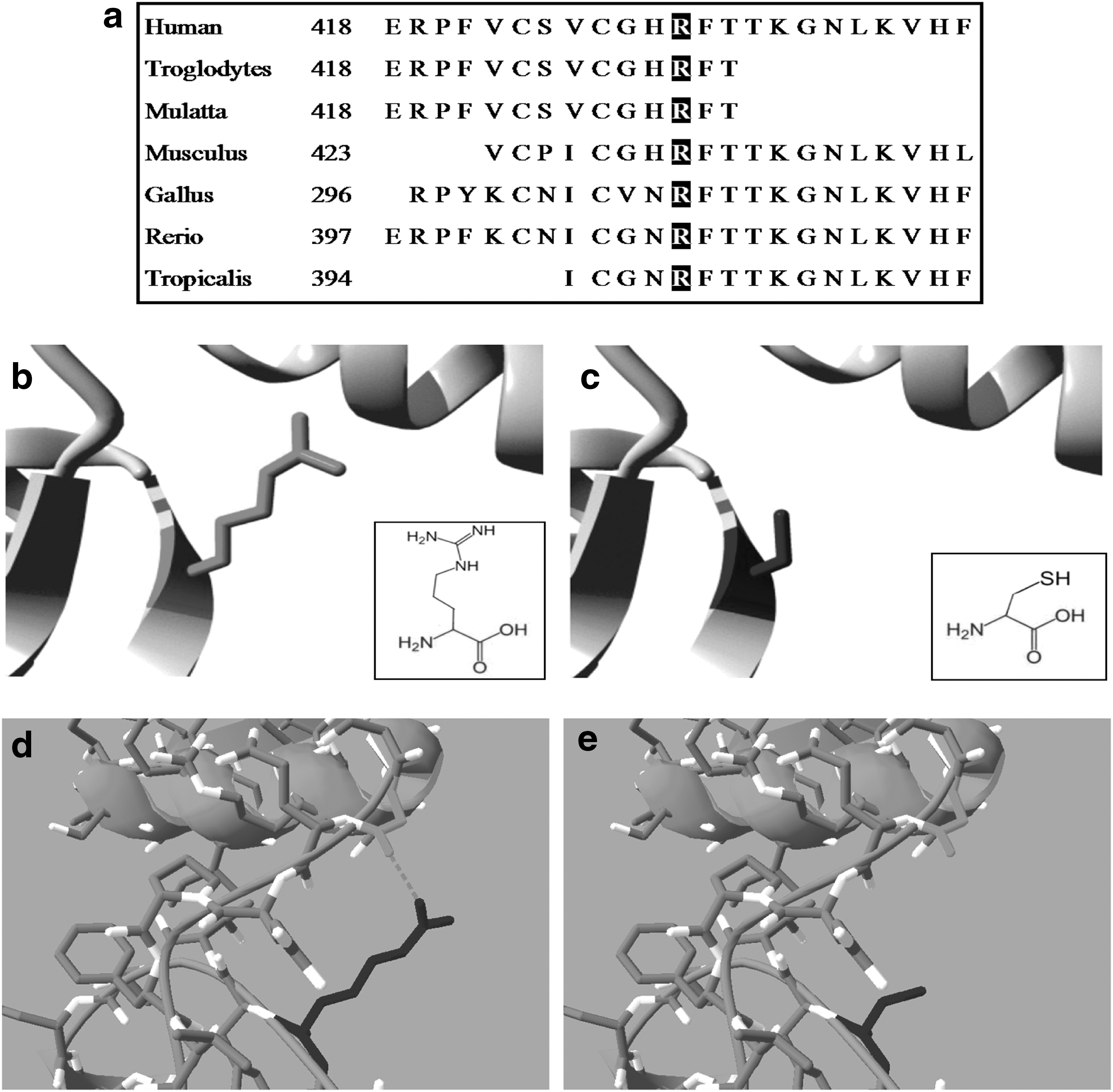
Biological analyses of SALL4-R418C mutation.
Discussion
In this study, we discovered a newborn HOS patient with multiple congenital malformations, including TOF, thumb agenesis, facial dysplasia, and right ear canal malformation. To clarify the cause, we conducted genetic analyses of the proband. We first used chromosomal microarray technology to check whether there were large fragments of chromosome duplication or deletion. Alterations of chromosome fragments usually implicate expression changes of several genes and lead to clinical abnormities of multiple organs or syndromes. While in our case, we did not detect any potential pathological alterations of chromosome fragments.
We next used WES to detect whether there were nucleotide changes of related genes. We conducted a preliminary quality filter of the data obtained and ruled out those low-quality sites that may be caused by sequencing technique. We eliminated those synonymous SNVs because they would not result in any functional changes of genes. Mutation sites were preferably located in the exon region, especially the coding sequence region, because mutations in these regions have higher risk to alter the function of the proteins and cause disease. We also screened the frequency of mutations and picked out those sites that may be rare mutations (max population frequency was <0.001; lower than rate of congenital heart disease). We found one HOS related gene SALL4 and picked out other five candidate genes contributing to syndromes or heart development or cardiac diseases. PLXDC1, SELE, and TTN, respectively, gives rise to endocardium disease, atherosclerosis, and cardiomyopathy (Mainardi et al., 1994; Zhao et al., 2014; Linschoten et al., 2017). However, mutations of these genes might not be related to the phenotypes in our case. KDM6A, KIF7, and SALL4 could lead to syndromes; KDM6A-induced kabuki syndrome is mainly manifested as congenital mental retardation accompanied with postnatal dwarfism and peculiar kabuki facies (Yang et al., 2016). KIF7 causes many syndromes among which the leading phenotypes are confined to developmental or functional abnormalities of nervous system; skeletal deformities are mainly characterized by polydactyly. Although Bardet–Biedl syndrome and Pallister–Hall syndrome show part phenotype of CHDs, CHDs in these two syndromes only act as concomitant symptoms rather than major roles (Putoux et al., 2011; Karaer et al., 2015; Krajewska-Walasek et al., 2015). According to our knowledge of the cause of disease accompanied with layer-by-layer screening of sequencing data, we thought that the mutations of SALL4 gene in this case were the most potential cause of our patient's disease.
To further analyze the association between SALL4 gene mutations in our study and HOS, we used several methods to discuss the pathological significance of the two candidate sites. Online software SIFT, PROVEAN, PolyPhen, and MutationTaster provided relatively consistent results: SALL4-M143V may be benign and plays as polymorphism site, while R418C could change protein function and cause disease. We also use web service HOPE to analyze the structural effects of SALL4 point mutation, especially, alterations of amino acid properties and domain function arising from mutant. HOPE indicated that SALL4-M143V can only give rise to amino acid size change; the effect of SALL4-R418C, however, can lead to changes in size, charge, and hydrophobicity between wild-type and mutant amino acid. What's more, mutated residue of SALL4-R418C is located in a Zinc figure domain of SALL4 protein, a domain that is important for binding of other molecules, domains, or DNA; mutation in this domain might disturb these interactions and impact the biological activities of SALL4 protein. HOPE also suggested that functional alterations of SALL4 protein may be due to mutations affecting the formation of hydrogen bonds. We then constructed the spatial conformation of SALL4 protein by SWISS PDB viewer and found that mutant cysteine disrupted the hydrogen bond connection between wild-type Arginine in position 418 and glycine in position 406. This output was in agreement with the result of HOPE; instability of protein and influenced binding ability were highly resulted from the loss of hydrogen bond in SALL4 Zinc figure domain. Based on the above analysis, we believe that the two screened SALL4 mutation sites, SALL4-M143V may be a mild mutant, and SALL4-R418C is likely to be a pathological locus and cause disease.
We diagnosed the patient as HOS because of her marked heart abnormalities and poor thumb development, but if we linked together congenital heart disease and another obvious phenotype, crooked mouth cry, it would be more reasonable to give the diagnosis of cardiac-facial syndrome, also known as DiGeorge syndrome. Facial abnormality of DiGeorge syndrome is mainly caused by facial dysplasia rather than facial paralysis, birth trauma, malpresentation, or other factors (Adeyinka et al., 2004; Chen et al., 2016). We could not rule out whether these factors play a role in the development of facial deformities in this newborn depending on current examinations. More importantly, genetic analyses found no 22q11 microdeletion or alterations of TBX1 gene which may be the leading cause of DiGeorge syndrome. Diagnosis of Okihiro syndrome, on the other hand, may be another confusion if SALL4 mutation was indeed the leading reason of the patient's disease. Okihiro syndrome is a SALL4-related syndrome, which is distinguished by radial malformations and Duane congenital eye movement disorder. Other phenotypes of Okihiro syndrome embrace anal stenosis, pigmentary disturbance, hearing deficit, renal malformations, ear malformations, facial asymmetry, and cardiac lesions, particularly atrial septal defect (Diehl et al., 2015; Alves et al., 2016). HOS and Okihiro syndrome cannot be distinguished by the phenotype heterogeneity of heart and upper limbs, while Okihiro syndrome exhibits Duane congenital eye movement disorder and visual disorder, which have been identified as the central clinical features. Confirmation of Duane symptoms relies on thorough ophthalmologic examinations (Kohlhase et al., 2003); we could not do eye movements and visual examinations since our patient was a newborn baby. However, both syndromes have the etiology of SALL4 mutation. Comparatively, it might not be the most crucial thing to give a precise diagnosis at this stage.
To discuss the genetic effect of the pathogenic mutation, we sequenced mutant sites of SALL4, KIF7, and TTN among family members. The father shared the same KIF7-R832W mutation with the proband and the mother shared SALL4-R418C and TTN-G27074R. That means, except from SALL4-M143V, the other mutations of the proband were all inherited from her parents while they had quite different phenotypes. Although our existing analyses based on online software suggested that SALL4-M143V may be tolerable, we could not completely deny that it has nothing to do with the occurrence of disease. We also speculated that the disease may be caused by cumulative effect of different genetic factors since the proband carried several pathological mutations; these mutations separately or cooperatively contribute to phenotypes of the proband. Finally, relationship of genotype and phenotype could be regulated by environmental factors; different environmental stimulation during pregnancy could cause genotypic phenotypic heterogeneity.
In summary, we preliminarily diagnosed a newborn baby with TOF, thumb agenesis, facial dysplasia, and right ear canal malformation as HOS. Using chromosome microarray and WES technology, we found a SALL4 gene mutation and predict that it could be a pathological site by bioinformatic analyses. Our study tended to deepen the understanding of phenotype difference among syndromes and role of SALL4 mutations in disease occurrence. While a more definitive diagnosis requires follow-up of the patient, the specific mechanism of genotype phenotype inconsistency also needs further exploration.
Footnotes
Acknowledgments
The study was supported by the National Natural Science Foundation of China (grant no. 81670210), Shanghai Municipal Commission of Health and Family Planning three-year action plan (GWIV-23), and Shanghai Jiao Tong University Medical Cross Fund (grant no. YG2015MS70).
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.