
Abstract
Macroautophagy, hereafter autophagy, is a catabolic process that is important for maintaining cellular homeostasis. It can also be used by cells to remove intracellular microbial pathogens. However, the studies on hepatitis C virus (HCV) in recent years indicated that this virus could regulate this cellular pathway and use it to enhance its replication. HCV could temporally control the autophagic flux and use the autophagic membranes for the assembly of its RNA replication complex. In this report, we will discuss the biogenesis of autophagosomes induced by HCV and how HCV uses this autophagic pathway for its RNA replication.
Introduction
H
In recent years, many reports had been published to show that HCV could induce autophagy to support its own replication [for reviews, see Wang and Ou (2015); Chan and Ou (2017)]. Autophagy is a catabolic process by which cells remove protein aggregates and damaged organelles for recycling. This process begins with the formation of crescent membrane structures known as phagophores (also called isolation membranes) in the cytosol. The membranes of phagophores will subsequently expand to form enclosed double-membrane vesicles known as autophagosomes. Autophagosomes mature by fusing with lysosomes to form autolysosomes, in which the cargos of autophagosomes are digested by lysosomal enzymes for recycling (Levine and Kroemer, 2008). Many factors that are required for the formation of autophagic vacuoles have been identified. These factors include more than 30 autophagy-related proteins (ATGs) and two ubiquitin-like conjugation systems. In one conjugation system, ATG12 is covalently conjugated to ATG5, which then forms a complex with ATG16. This ATG5–ATG12–ATG16 complex is essential for the formation of phagophores, and it is dissociated from autophagosomes once they are formed (Xie and Klionsky, 2007; Geng and Klionsky, 2008). In the second conjugation system, the microtubule-associated light chain 3 (LC3) is covalently linked to the lipid phosphatidylenthanolamine. The lipidated LC3 is required for the formation of autophagosomes and, due to its association with autophagosomes, often used as the marker for these membrane vesicles (Rubinsztein et al., 2009).
Autophagy is ongoing in cells at a basal level to maintain cellular homeostasis. However, it can also be induced by stressors such as nutrient starvation or by treatment with pharmacological agents. It can also be used by cells to remove intracellular microbial pathogens in a process known as xenophagy (Levine, 2005). However, many microbial pathogens including HCV have developed mechanisms to subvert this intracellular antimicrobial pathway and use it to support their own replications (Dreux et al., 2009; Sir and Ou, 2010; Lennemann and Coyne, 2015).
Temporal Regulation of Autophagic Flux by HCV
HCV induced autophagic vacuoles in cells to enhance its own replication (Ait-Goughoulte et al., 2008; Sir et al., 2008). Our previous results indicated that the fusion between autophagosomes and lysosomes was inefficient in the early stage of HCV infection before the peak of viral release (Wang et al., 2015). This led to the accumulation of autophagosomes (Sir et al., 2008, 2012). This inhibition of autophagic flux was due to the induction of RUBICON by HCV. RUBICON is a RUN-domain BECLIN 1-interacting and cysteine-rich containing protein. It inhibits the fusion between autophagosomes and lysosomes by sequestering UVRAG from the homotypic fusion and protein sorting complex (Matsunaga et al., 2009; Zhong et al., 2009; Sun et al., 2010). The inhibitory effect of RUBICON was overcome by UVRAG, which was induced by HCV in the late stage of infection, resulting in the completion of the autophagic flux (Wang et al., 2015). The accumulation of autophagosomes in the early stage of HCV infection enhanced HCV RNA replication, as they provided membrane platforms for the assembly of the HCV RNA replication complex (Sir et al., 2008, 2012). The HCV RNA replication complex is associated with lipid rafts (Aizaki et al., 2004; Mizushima et al., 2008). These lipid rafts are also detected on autophagosomes in HCV-infected cells but not on autophagosomes induced by nutrient starvation or rapamycin, which inhibits mTOR complex I to induce autophagy (Kim et al., 2017). The completion of the autophagic flux in the later stage of the HCV life cycle allowed HCV to deplete TRAF6 via autophagy (Chan et al., 2016; Chan and Ou, 2017). TRAF6 is an important adaptor molecule for the production of interferons and proinflammatory cytokines. Thus, the temporal regulation of autophagic flux is very important for HCV, as it allows HCV to accumulate autophagosomes in the cell in the early stage of its life cycle to maximize its RNA replication and in the meantime use autophagic vacuoles to sequester and deplete TRAF6 to suppress the host innate immune responses.
Biogenesis of Autophagosomes Induced by HCV
Autophagosomal membranes may be derived from different subcellular organelles. It may be generated from endoplasmic reticulum (ER) membranes (Axe et al., 2008; Hayashi-Nishino et al., 2009; Yla-Anttila et al., 2009), outer membrane of mitochondria (Hailey et al., 2010), membranes of Golgi (Yen et al., 2010), early endosomes (Longatti et al., 2012), plasma membranes (Ravikumar et al., 2010), and vesicles budding from ER and Golgi (Zoppino et al., 2010; Guo et al., 2012). The ER–mitochondria junction (Hamasaki et al., 2013) and ER–Golgi intermediate compartment (Ge et al., 2013) were also found to be involved in the biogenesis of autophagosomes. It is possible that in different cell types, phagophores may originate from different subcellular compartments and, even within the same cell, the origin of phagophores may vary depending on the external stimuli. By using ATG5 as the marker for phagophores and by conducting live-cell imaging, we recently demonstrated that phagophores induced by HCV were derived from the ER membranes and could subsequently progress to become autophagosomes (Wang et al., 2017). Interestingly, the progression of phagophores to autophagosomes in nutrient-starved cells took ∼10 min whereas that induced by HCV took ∼30 min. This difference suggested the possible involvement of different molecular pathways in the biogenesis of autophagosomes induced by HCV and nutrient starvation.
Phagophores were previously thought to form autophagosomes through the elongation of their membranes while being encircled by their associated ER. Their membrane edges are eventually sealed to form the enclosed autophagosomes (Hayashi-Nishino et al., 2009; Tooze and Yoshimori, 2010). More recently, it was discovered that phagophores could also undergo homotypic fusion in a syntaxin 7 (STX7)-dependent manner to generate autophagosomes (Moreau et al., 2011). STX7 belongs to a family of soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) proteins. SNAREs on different membranes form helix bundles with their partners to mediate vesicle fusion (Schafer et al., 2012). It is possible that phagophores may undergo either elongation or homotypic fusion to form autophagosomes depending on the cells or stress conditions. In our recent studies, we found that autophagosomes induced by HCV were primarily generated via the homotypic fusion of phagophores (Wang et al., 2017). This conclusion was made based on the following observations: first, the electron microscopy analysis of cells that contained an HCV subgenomic RNA replicon revealed juxtaposed phagophores that were being assembling into a membrane vesicle that resembled autophagosomes; second, phagophores separately labeled with mCherry-ATG5 or mEmerald-ATG5 in HCV replicon cells or HCV-infected cells could undergo homotypic fusion in vitro; and third, the suppression of STX7 expression blocked the formation of autophagosomes without affecting phagophores in HCV replicon cells and HCV-infected cells.
Phagophores As the Initial Sites for the Assembly of HCV RNA Replication Complex
In our subsequent studies, we found that, besides autophagosomes, phagophores purified from HCV-infected cells could also mediate HCV RNA replication in vitro. In addition, the suppression of STX7 expression, which inhibited the formation of autophagosomes, did not suppress HCV RNA replication but rather, slightly increased it (Wang et al., 2017). These results indicated that the HCV RNA replication complex was assembled on phagophores and remained associated with autophagosomes. This could explain why the knockdown of STX7, which suppressed the homotypic fusion of phagophores and the formation of autophagosomes, had no effect on HCV RNA replication. Recently, it was reported that the knockdown of ATG12, which was required for the formation of phagophores, suppressed the HCV RNA replication (Fahmy and Labonte, 2017). This report lends further support to the argument that phagophores are the initial sites for the assembly of HCV RNA replication complex.
Conclusion
Recent studies provided significant insight for understanding how HCV regulates the autophagic pathway for its replication. As illustrated in Figure 1, phagophores induced by HCV are derived from the ER and serve as the assembly site for the HCV RNA replication complex. Phagophores induced by HCV then undergo homotypic fusion in an STX7-dependent manner to generate autophagosomes. The HCV RNA replication complex remains associated with autophagosomes after they are generated. The maturation of autophagosomes is suppressed by RUBICON, which is induced by HCV. This leads to the accumulation of autophagosomes, which support HCV RNA replication. At the later stage of the HCV life cycle, UVRAG was induced to antagonize the activity of RUBICON, leading to the fusion of autophagosomes and lysosomes to form autolysosomes. The studies on HCV provide important information for understanding how a virus may control the autophagic pathway to benefit its own replication.
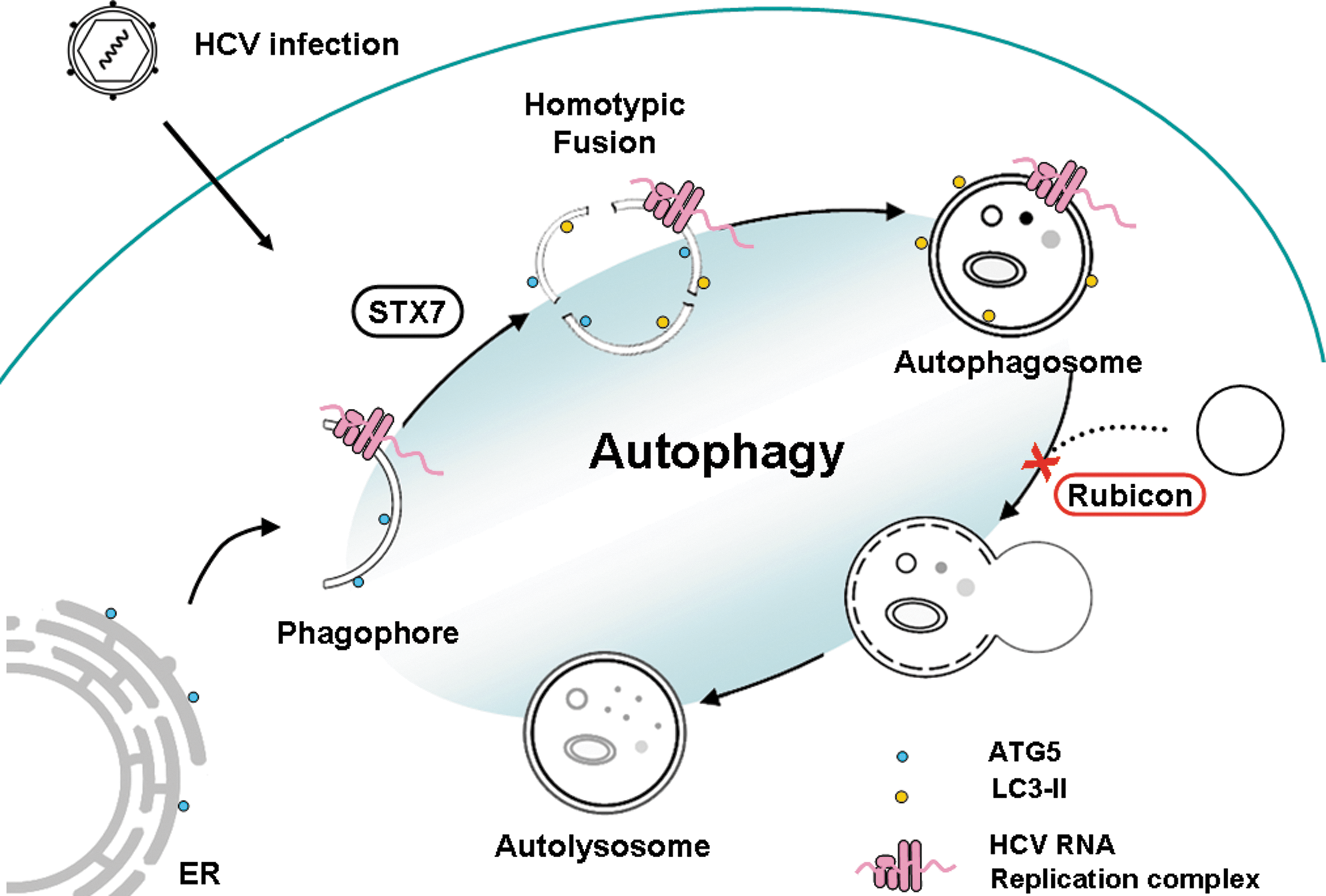
Illustration of how HCV regulates autophagy. HCV infection induces the localization of ATG5 to the ER and the subsequent formation of phagophores. Phagophores undergo homotypic fusion in a STX7-dependent manner to form autophagosomes. Both phagophores and autophagosomes can support HCV RNA replication. HCV induces RUBICON expression in the early stage of its life cycle to block the fusion of autophagosomes and lysosomes, resulting in the accumulation of autophagosomes that support HCV RNA replication. HCV, hepatitis C virus; ER, endoplasmic reticulum; ATG, autophagy-related protein; STX7, syntaxin 7.
Footnotes
Acknowledgment
This work is supported by National Institutes of Health grant DK094652.
Disclosure Statement
No competing financial interests exist.