
Abstract
Mitochondria are critical for cellular survival, and for their proper functioning, translocation of ∼1500 proteins across the mitochondrial membranes is required. The translocase of the outer (TOMM) and inner mitochondrial membrane (TIMM) complexes are major components of this translocation machinery. Through specific processes, preproteins and other molecules are imported, translocated, and directed to specific mitochondrial compartments for their function. In this study, we review the association of subunits of these complexes with human disease. Pathogenic mutations have been identified in the TIMM8A (DDP) and DNAJC19 (TIMM14) genes and are linked to Mohr-Tranebjærg syndrome and dilated cardiomyopathy syndrome (with and without ataxia), respectively. Polymorphisms in TOMM40 have been associated with Alzheimer's disease, frontotemporal lobar degeneration, Parkinson's disease with dementia, dementia with Lewy bodies, nonpathological cognitive aging, and various cardiovascular-related traits. Furthermore, reduced protein expression levels of several complex subunits have been associated with Parkinson's disease, Meniere's disease, and cardiovascular disorders. However, increased mRNA and protein levels of complex subunits are found in cancers. This review highlights the importance of the mitochondrial import machinery in human disease and stresses the need for further studies. Ultimately, this knowledge may prove to be critical for the development of therapeutic modalities for these conditions.
Introduction
M
In humans, the mitochondrial proteome consists of roughly 1500 different proteins and possesses its own genome and protein synthesis machinery (Pagliarini et al., 2008). Approximately 1% of mitochondrial proteins is encoded by the mitochondrial genome, whereas the remaining 99% are encoded as precursor proteins by nuclear genes, synthesized in the cytosol on ribosomes and imported into the mitochondria through complex processes (Endo et al., 2003; Wiedemann et al., 2004; Straub et al., 2016). These import processes rely on the assistance of cytosolic chaperone molecules such as heat shock proteins 70 and 90 (HSP70 and HSP90).
Preproteins and other molecules are imported, translocated, and directed to specific mitochondrial compartments, including the outer mitochondrial membrane (OMM), the intermembrane space (IMS), the inner mitochondrial membrane (IMM), or the matrix, based on specific mitochondrial target sequences. Mitochondrial import and translocation mechanisms have previously been reviewed in great depth (MacKenzie and Payne, 2007; Endo and Yamano, 2009; Harbauer et al., 2014; MacPherson and Tokatlidis, 2017; Kang et al., 2018). Hence, for contextual purposes, only a brief overview of the protein complexes and processes involved will be provided.
The translocase of the outer mitochondrial membrane (TOMM) complex (Fig. 1) represents the central entry gateway for almost all mitochondrial proteins (Ryan et al., 2000; Dolezal et al., 2006; Chacinska et al., 2009). TOMM20, TOMM22, and TOMM70 function as the main receptors on the mitochondrial surface with their hydrophilic domains exposed to the cytosol and their transmembrane N-terminal domains acting as anchors. TOMM70 and TOMM20 have similar functions, but recognize different precursor proteins. TOMM20 is an important receptor subunit of the complex and serves by recognizing mitochondrial precursor proteins with cleavable N-terminal presequences (Ellenrieder et al., 2015; Johnson et al., 2015). This subunit also allows for mitochondrial localization of nuclear-encoded mitochondrial oxidative phosphorylation subunits (Johnson et al., 2015). TOMM70 shows a high affinity for proteins with an internal hydrophobic targeting sequence such as carrier proteins (Endo et al., 2003; Wu and Sha, 2006; Yamano et al., 2008; Shiota et al., 2011; Ellenrieder et al., 2015). TOMM22 also recognizes precursor proteins with N-terminal presequences and connects TOMM20 to the pore of the complex known as TOMM40. In addition, the IMS domain of TOMM22 binds and guides precursor proteins to other translocases such as the translocase of the inner mitochondria membrane (TIMM) import complexes (Van Wilpe et al., 1999; Yamano et al., 2008; Shiota et al., 2011).
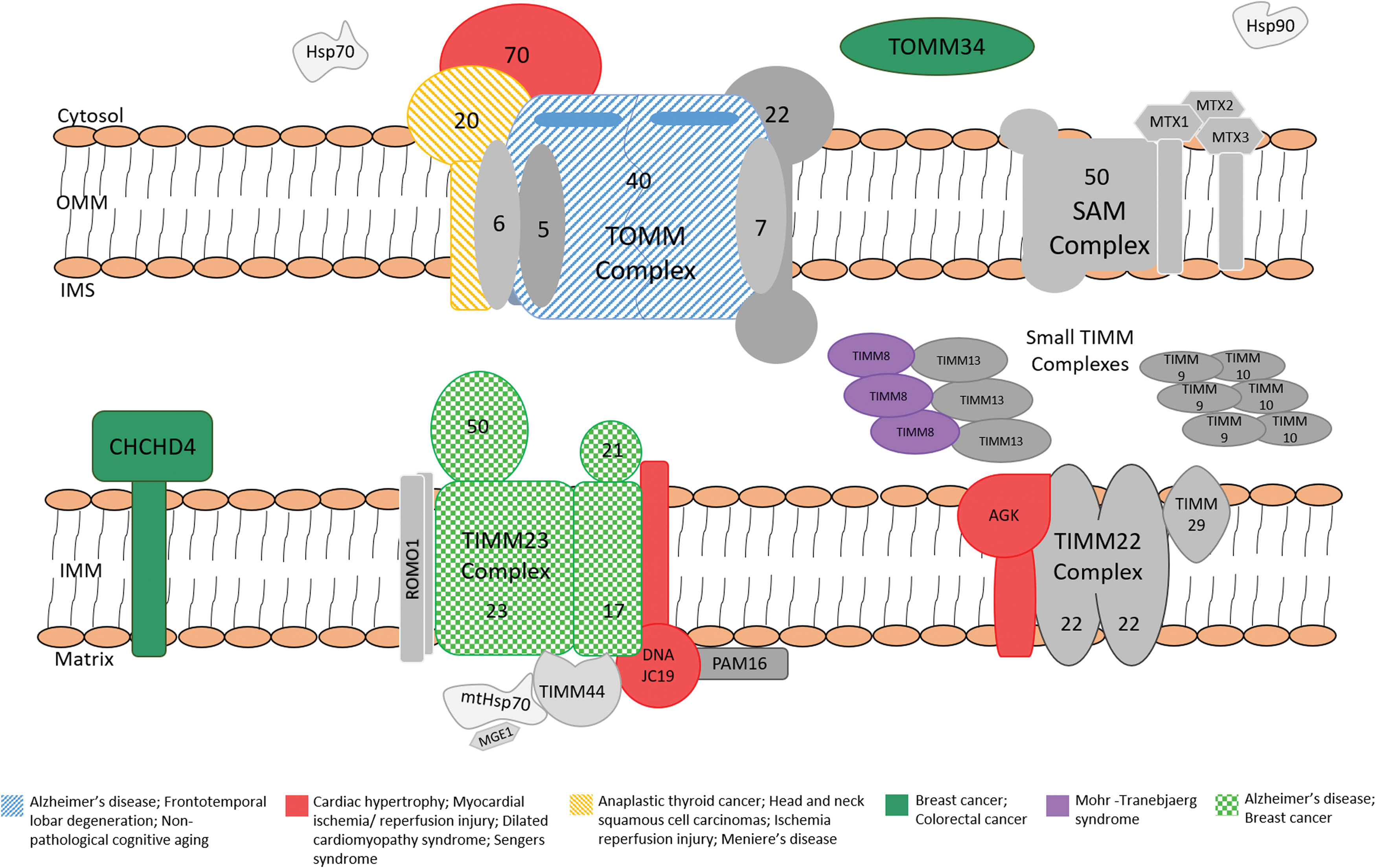
Components of the mitochondrial protein import machineries and their association with human disease. Mitochondrial proteins are synthesized in the cytoplasm and imported through the translocase of outer mitochondrial membrane (TOMM) complex. The subsequent sorting of proteins to their final destination requires the action of additional import complexes. Proteins containing a presequence are transferred from the TOMM complex to the translocase of the inner mitochondria membrane (TIMM23) complex that can translocate proteins into the matrix or the inner membrane. Precursor proteins of inner membrane are translocated to the TIMM22 complex, which mediates their integration into the inner membrane. The small proteins that reside in the IMS are recognized by the MIA machinery (not shown) on their exit from the TOM complex. Finally, β-barrel precursors are relayed from the TOMM complex to the sorting and assembly machinery complex, which facilitates their assembly into the outer membrane. For clarity, only the components of interest to this review have been indicated in this illustration. IMS, intermembrane space; MIA, mitochondrial IMS assembly. Color images available online at
TOMM40, the central component of the TOMM complex that forms the protein-conducting channel to transport precursor proteins across the outer membrane, is essential for cell viability (Pfanner et al., 2004; Ellenrieder et al., 2015). TOMM40 also serves as a “sophisticated sorting station” and is involved in sorting substrates to various submitochondrial compartments (Gabriel et al., 2003). Molecular chaperones HSP70 and HSP90 are important for targeting preproteins to the TOMM complex and protect these proteins from aggregating in the cytosol (Young et al., 2003). The membrane proteins are delivered to the mitochondria by docking with TOMM70, the import receptor (Faou and Hoogenraad, 2012). TOMM34 was recently discovered and described as a scaffold co-chaperone of HSP70/HSP90, forming an HSP70-TOMM34-HSP90 tripartite complex, which aids in precursor protein import by maintaining the precursor proteins in translocation-competent conformations, and eventually for folding into their native conformations following translocation (Faou and Hoogenraad, 2012; Trcka et al., 2014).
Following translocation through the TOMM complex, the mitochondrial proteins separate into (i) the sorting and assembly machinery (SAM), (ii) the translocase of IMM complexes (TIMM22 and TIMM23), or (iii) the mitochondrial IMS assembly (MIA) pathway (Wiedemann et al., 2004; MacKenzie and Payne, 2007; Stojanovski et al., 2012). These are complex pathways that require the correct “signals” to function properly. For example, proteins of the carrier family with an internal hydrophobic targeting sequence are transferred to small TIMM chaperones in the IMS and guided to TIMM22, which is responsible for translocating proteins in the inner membrane. The TIMM22 complex consists of different subunits. The core component of the complex is TIMM22, which mediates insertion of carriers in the inner membrane in a membrane potential manner. TIMM54 is another TIMM22 subunit that might function as a receptor for small TIMM chaperones, whereas TIMM18 is important for stability and assembly of the translocase, but these subunits have only been identified in yeast. TIMM29 plays an important role in the assembly of the TIMM22 complex (Callegari et al., 2016; Kang et al., 2016), and recently, acylglycerol kinase (AGK) was also identified as a subunit of TIMM22 (Kang et al., 2017; Vukotic et al., 2017) (Fig. 1). AGK was shown to function in a kinase-independent manner to maintain the integrity of the TIMM22 complex, where it facilitates the import and assembly of mitochondrial carrier proteins.
The TIMM23 complex is necessary for directing proteins into either the inner membrane or matrix, depending on their signal (Chacinska et al., 2009). Precursor recognition and interaction with the TOMM complex are facilitated by IMS domains of the subunits TIMM23, TIMM50, and TIMM21 (Lytovchenko et al., 2013). The translocation channel is formed by TIMM23 together with TIMM17 (van der Laan et al., 2007). The complex is connected to members of the respiratory chain complex by TIMM21. Thereby, protein transport and the generation of membrane potential are coupled, which is important for the insertion of inner membrane proteins and translocation of proteins in the matrix (van der Laan et al., 2006). Other proteins associated with the TIMM23 complex are the reactive oxygen species (ROS) modulator 1, responsible for regulating mitochondrial ROS production and acts as a redox sensor in mitochondrial dynamics (Chung et al., 2006; Lee et al., 2018), and PAM16 (MAGMAS), which is needed for the translocation of preproteins into the matrix, but is not required for protein insertion into the inner membrane (Frazier et al., 2004; Sinha et al., 2010). The core component of the motor consists of mtHSP70 and is driven by ATP binding and hydrolysis. This ATP/adenosine diphosphate (ADP) exchange is stimulated by the nucleotide exchange factor Mge1 and the ATPase activity is enhanced by TIMM14. The preproteins that are processed by the TIMM23 complex contain N-terminal presequences that are removed by the mitochondrial processing peptidase (Jensen and Dunn, 2002; Ieva et al., 2013).
The SAM complex is responsible for the insertion of β-barrel proteins into the outer membrane with the assistance of a family of metaxin proteins (MTX1, MTX2, and MTX3), which is proposed to be required for the biogenesis of these β-barrel proteins (Chan and Lithgow, 2008; Huynen et al., 2016; Kang et al., 2018). Another outer membrane complex, the mitochondrial import machinery (MIM), promotes the import and insertion of α-helical outer membrane proteins (Becker et al., 2008b). However, to date, this complex has only been identified in yeast, but not in humans.
IMS proteins with incorporated cysteine motifs are recognized by CHCHD4 (MIA40), the core component of the MIA machinery, which promotes formation of disulfide bonds in the precursor proteins (Stojanovski et al., 2012; Ceh-Pavia et al., 2013). These disulfide bonds stabilize the folded state of the proteins, which keep them trapped in the IMS (Straub et al., 2016). CHCHD4 has also been found to be highly conserved and is incorporated into the IMM by TIMM23.
Most α-helical outer membrane proteins are directly inserted into the target membrane from the cytosol, whereas a portion of these proteins, including multispanning membrane proteins, is targeted to mitochondria and recognized by TOMM70 (Ellenrieder et al., 2015). These multispanning membrane proteins are inserted into the OMM with the assistance of the MIM machinery (Becker et al., 2008a, 2011; Hulett et al., 2008; Popov-Čeleketić et al., 2008; Papić et al., 2011; Dimmer et al., 2012; Ellenrieder et al., 2015; Wenz et al., 2015); however, the exact translocation process remains unclear.
Since mitochondria are known to be essential for cellular survival, defective mitochondrial import and assembly complexes are expected to have a major impact on human health and well-being. However, the roles of these complexes in human disease have not been well studied. This review summarizes the current knowledge on defects (with a focus on genetic alterations and protein expression levels) in mitochondrial protein import machineries in various disorders and highlights the need for further investigation in this understudied field. In light of the available published research, this review will focus mainly on the role of the protein import complexes, TOMM and TIMM, and how they have been implicated in the development of multiple diseases. These studies are summarized in Tables 1 and 2.
Association of Genetic Variants in the Translocase of the Outer Mitochondrial Membrane and Translocase of the Inner Mitochondria Membrane Subunits with Human Diseases
AD, Alzheimer's disease; CRC, colorectal cancer; DCMA, dilated cardiomyopathy with ataxia syndrome; FTLD, frontotemporal lobar degeneration; HNSCC, head and neck squamous cell carcinomas; LOAD, late-onset AD; MTS, Mohr-Tranebjærg syndrome; N/A, not applicable; PD, Parkinson's disease; PDD, Parkinson's disease with dementia, DLB, dementia with Lewy bodies; AGK, acylglycerol kinase; APOE, apolipoprotein E allele; MTS, Mohr-Tranebjærg syndrome; GAC, Guanine, Adenine, Cytosine; SNP, single nucleotide polymorphism.
Altered Gene and Protein Expression Levels of Translocase of the Outer Mitochondrial Membrane and Translocase of the Inner Mitochondria Membrane Subunits Associated with Human Diseases
MD, Meniere's disease; Aβ, amyloid-β peptide; APP, amyloid precursor protein; α-synuclein, alpha-synuclein.
Involvement in Human Disease
Neurological disorders
Alzheimer's disease
Alzheimer's disease (AD) is the most common neurodegenerative disorder and patients with this condition suffer from a decline in memory and cognition, as well as in various activities of daily living. Histopathological analyses on brains of AD patients have revealed two major molecular hallmarks: intracellular fibrillar tangles mainly composed of hyperphosphorylated tau protein and extracellular neuritic plaques, which predominantly comprises amyloid-β peptide (Aβ). While it is well established that AD is a multifactorial disorder, it has been noted that mitochondrial dysfunction is among the earliest observed pathogenic alterations, and it is detected well before the accumulation of neuritic plaques occurs (Yao et al., 2009).
Aβ peptide is derived from the amyloid precursor protein (APP), which can be processed by various proteases to generate different Aβ species (Pinho et al., 2014). Of these, Aβ40 is the most abundant, but the most toxic is Aβ42 due to its higher hydrophobicity and the concomitant higher propensity to aggregate (Haass and Selkoe, 2007).
AD has been categorized broadly into two main subtypes: the autosomal dominant early-onset form with age at onset (AAO) before 60 years of age, and the “sporadic” or late-onset AD (LOAD) where disease presentation is typically >60 years of age. The strongest genetic risk factor for development of LOAD is the apolipoprotein E allele ɛ4 (APOE ɛ4), which is associated with a lower age of clinical disease onset (Corder et al., 1993; Grossman et al., 2010). The APOE gene is located on chromosome 19 and it resides in a region of linkage disequilibrium (LD) that includes the TOMM40, apolipoprotein C1, and nectin cell adhesion molecule 2 (NECTIN2/PVRL2) genes (Lutz et al., 2010).
Mutations/susceptibility factors in TOMM40
The TOMM40 gene has been associated with the AAO of LOAD in several genetic association studies. One of the polymorphisms in TOMM40 is rs10524523, which is located in intron 6 and is composed of thymidine (T) repeats varying between 11 and 43 nucleotides in length (Linnertz et al., 2012). Three categories of alleles have been described: short (S, T ≤ 19 nucleotides), long (L, 20 ≤ T ≤ 29) nucleotides), or very long (VL, T ≥ 30 nucleotides) (Lutz et al., 2010).
It has been speculated that the discovery of APOE ɛ4 as a genetic risk factor for LOAD may have occurred because this allele is almost always linked to the long allele of rs10524523 in TOMM40 (∼98% of the time) (Lutz et al., 2010). Notably, the co-occurrence of ɛ4 with the L allele occurs mostly in the Caucasian population, whereas in African Americans, a significant number of ɛ4 alleles occur in individuals with the S/S genotype (Linnertz et al., 2012). Phylogenetic analyses have revealed that the common ɛ3 allele has a separate genetic origin and that it is linked to either S or VL poly-T variants, which challenges the commonly accepted hypothesis that the ɛ3 allele is AD risk-neutral (Grossman et al., 2010).
A number of studies attempted, but were not able to replicate the association of TOMM40 rs10524523 with AAO of LOAD, as reviewed previously (Roses et al., 2013). Various methodological and technical problems could account for these varying results, which may include difficulties with accurately determining the length of the alleles and the lack of standardized determination of the allele categories and of the AAO in LOAD (Roses et al., 2013).
Interestingly, it has been reported that polymorphisms in TOMM40 may not have an APOE-independent role in the risk of developing LOAD and frontotemporal lobar degeneration (FTLD). In one study in which three TOMM40 single nucleotide polymorphisms (SNPs) (rs157580, rs2075650, and rs157581) were genotyped, an association with these SNPs was found for LOAD, but not FTLD (Bagnoli et al., 2013). However, when the presence of the APOE allele was taken into account, a strong association between the ɛ4 allele and the Guanine Adenine Cytosine (GAC) haplotype for the three SNPs was identified in both LOAD and FTLD patients.
mRNA and protein expression levels of TOMM40
It has been shown that the mRNA expression of TOMM40 and APOE is significantly increased in brain tissue from LOAD cases compared to controls (Linnertz et al., 2014). Interestingly, the mean expression of mRNA levels of both genes was higher in rs10524523 VL homozygotes compared with S homozygotes in the temporal and occipital cortexes. To test the effect of rs10524523 in a genomic context, a luciferase reporter system containing an ∼7 kb genomic fragment was generated in two human cell lines (HepG2 hepatoma and SH-SY5Y neuroblastoma cells). The results were consistent with the human brain mRNA analysis; the VL allele resulted in significantly higher TOMM40 expression than the S allele. Although the effect was the same in both cell lines, the magnitude of the effect was greater in the neuroblastoma than in the hepatoma cells, which implies tissue-specific regulation of the rs10524523 poly-T. However, contrasting findings were obtained from a study conducted on dermal fibroblasts from cognitively healthy APOE ɛ3/ɛ4 carriers, which revealed that TOMM40 rs10524523 alleles do not affect TOMM40 mRNA or protein levels or the size of TOMM40 protein (Hedskog et al., 2012).
In the cortex of C57BL/6J mice, metformin (a drug used for the treatment of diabetes) increases the protein levels of TOMM40, resulting in mitochondrial dysfunction and cell death (Picone et al., 2016). Metformin also increases the levels of APP, thereby promoting the aggregation of Aβ predominantly in the cortex region. These observations suggest a strong association between metformin-induced Aβ accumulation and mitochondrial dysfunction through an impaired TOMM40 expression that results in mitochondrial stress and, in turn, a source of increased Aβ production. However, due to the complex interactions between these different factors within this cascade, it is difficult to establish a clear sequence of events.
Accumulation of APP in TOMM/TIMM complexes
Abnormal accumulation of APP across TOMM and TIMM import channels could be considered a hallmark of AD pathology. Devi et al. found nonglycosylated full-length and C-terminal truncated APP that accumulated specifically in mitochondrial import channels in postmortem AD brains, but not in nondemented controls (Devi et al., 2006). In AD brains specifically, APP formed stable complexes with TOMM40, and supercomplexes with both TOMM40 and TIMM23. This accumulation of incompletely translocated APP blocked the transport of nuclear-encoded proteins leading to dysfunctional mitochondria. Notably, patients with APOE ɛ3/ɛ4 alleles were found to have the highest content of mitochondrial APP. Aβ has also been shown to be translocated into the mitochondria through the TOMM machinery (Hansson Petersen et al., 2008). Following import, Aβ is inserted into the mitochondrial inner membrane and it was also found in the matrix. However, the function of Aβ in the mitochondria and how it gains access to the different mitochondrial subcompartments remain to be elucidated (Pinho et al., 2014).
Alterations in mitochondrial function, morphology, and dynamics associated with Aβ accumulation may be one of the earliest alterations in AD pathology (Yao et al., 2009; Pinho et al., 2014). Although it is known that this peptide is imported into mitochondria by the TOMM machinery, understanding how Aβ reaches the mitochondria (possibly through synthesis from APP accumulated within the translocation channels) and how its mitochondrial levels are regulated may provide important clues to the pathobiology of AD. Presequence protease (PreP) is localized in the mitochondrial matrix and is responsible for degrading mitochondrial targeting sequences when the protein reaches its correct destination following import. Human PreP can degrade Aβ, and significantly lower levels of hPreP have been found in the brain temporal lobe (a region highly susceptible to Aβ accumulation) of AD patients compared to controls (Pinho et al., 2014).
Furthermore, in AD brains, it has been shown that Aβ impairs maturation of mitochondrial preproteins causing an accumulation of nonprocessed mitochondrial preproteins and processing intermediates (Mossmann et al., 2014). It does this by inhibiting the degradation of presequence peptides by PreP, and this eventually leads to accelerated protein degradation and an imbalance of the mitochondrial proteome. Taken together, these findings suggest that manipulation of Aβ import and accumulation in mitochondria could potentially be a therapeutic target to alleviate AD symptoms.
Accumulation of anti-TOMM40 antibodies
Significantly higher levels of anti-TOMM40 antibodies have been noted in the serum of AD patients compared to control subjects or patients with multiple sclerosis (Kimura et al., 2012). Also, cognitive impairment, as measured by the Mini-Mental State Examination, was significantly associated with the presence of anti-TOMM40 antibodies in AD patients. The mechanism of anti-TOMM40 antibody production is unclear, but the authors speculate that the antibody may penetrate cell membranes and bind to TOMM40, and this may inhibit protein import into mitochondria causing mitochondrial dysfunction.
Parkinson's disease
Genetic association with variants in TOMM40
Parkinson's disease with dementia (PDD) and dementia with Lewy bodies (DLB) are two forms of PD that are associated with dementia and share common clinical and pathological features. The L allele of TOMM40 rs10524523 was found to be significantly associated with PDD/DLB patients compared to PD patients without dementia (Lindqvist et al., 2016). However, when adjusting for APOE ɛ4 allele status, this association was no longer significant. To determine whether the association may be mediated by increased AD-related pathology, cerebrospinal fluid levels of Aβ42 and tau were measured in a subpopulation of patients; the L allele was significantly associated with lower Aβ42 as well as lower Aβ42/tau levels in all patients. However, again these findings were not significant when adjusting for the APOE ɛ4 allele status. The authors conclude that the rs10524523 polymorphism's effects on PDD and DLB pathology may be driven by APOE ɛ4, but that that these findings need to be replicated and validated in independent patient cohorts.
Alpha-synuclein and TOMM proteins
Alpha-synuclein (α-synuclein) is a protein implicated in PD, and aggregation of this protein in the brains of PD patients, in structures known as Lewy bodies, is a key feature of the neuropathology (Spillantini et al., 1997). TOMM40 protein levels are significantly decreased in postmortem brains of PD patients and in wild-type human α-synuclein transgenic mice, whereas TOMM20 protein levels remain unchanged (Bender et al., 2013). In α-synuclein transgenic mice, the reduced TOMM40 levels were associated with increased deletions in mitochondrial DNA (mtDNA) and oxidative DNA damage, and with decreased energy production and altered levels of complex I proteins. Interestingly, these effects were rescued in α-synuclein transgenic mice brains using a lentiviral-mediated overexpression of Tomm40. These results suggest that alterations in the mitochondrial protein transport machinery might contribute to mitochondrial impairment in α-synucleinopathies.
Certain post-translationally modified species of α-synuclein bind specifically to TOMM20, preventing its interaction with TOMM22, thereby impairing mitochondrial protein import (Di Maio et al., 2016). This ultimately leads to reduced respiration and excessive ROS production. The interaction was not observed between α-synuclein and TOMM22, TOMM40, or TIMM23. Notably, overexpression of TOMM20 was found to ameliorate the deleterious effect of α-synuclein on protein import, showing promise for potential therapeutic intervention.
PINK1-PRKN pathway and interaction with TOMM proteins
Loss-of-function mutations in the PRKN (PARK2) and PINK1 genes are implicated in early-onset autosomal recessive forms of PD (Corti et al., 2011). PINK1 and PRKN work together in a pathway to clear dysfunctional mitochondria by the process of mitophagy. When a mitochondrion sustains damage that leads to a loss in membrane potential by uncouplers or oxidative stress, PINK1 accumulates on the OMM, resulting in PINK1-dependent recruitment and activation of PRKN, which ultimately trigger selective degradation of the faulty mitochondrion (Narendra et al., 2008). PINK1 uses a unique mitochondrial localization mechanism that may work together with the TOMM complex, specifically TOMM40, to promote localization to the OMM (Okatsu et al., 2015).
Following mitochondrial membrane potential loss, PINK1 forms a 700 kDa complex on the OMM with various TOMM subunits, including TOMM70, TOMM22, TOMM20, and TOMM40 (Lazarou et al., 2012). Interestingly, blockage of mitochondrial protein import can trigger the recruitment of PRKN, by PINK1, to the TOMM machinery (Bertolin et al., 2013). PD-causing PRKN mutations can weaken or disrupt the interaction between PRKN and TOMM70, and TOMM40. Overproduction of TOMM22 or TOMM40 reverses mitochondrial clearance promoted by PINK1 and PRKN following loss of membrane potential. These results reveal that the TOMM machinery plays an integral role in the mitochondrial clearance program controlled by the PINK1-PRKN pathway by serving as “a key molecular switch” (Bertolin et al., 2013).
A matrix protein substrate of PRKN, 17-β hydroxysteroid dehydrogenase type 10 (HSD17B10), can recruit PRKN to the TOMM complex, in close proximity to PINK1, in healthy and depolarized mitochondria (Bertolin et al., 2015). However, PRKN can still be recruited to mitochondria in cells lacking HSD17B10, indicating that it does not play a vital role in this process. The authors speculate that maintenance of appropriate levels of mitochondrial HSD17B10, by the TOMM machinery, is a mechanism whereby PRKN (possibly in collaboration with PINK1) preserves mitochondrial quality. Loss of this protective mechanism may lead to mitochondrial dysfunction and neuronal death. In the event that mitochondria are able to reestablish the mitochondrial membrane potential, PINK1 accumulated on the OMM is readily reimported and degraded (Lazarou et al., 2012). This then leads to deactivation of PRKN, termination of mitophagy, and maintenance of healthy organelles in the network.
Nonpathological cognitive aging
Variants in TOMM40 were associated with nonpathological cognitive aging in the first genome-wide association study (GWAS) conducted on this phenotype (Davies et al., 2014). In this study, five UK cohorts of older adults with longitudinal cognitive ability data were used as the discovery cohort and three independent Swedish cohorts (from the Swedish Twin registry) were used as the replication cohort. All study participants were living independently in the community and none was diagnosed with AD or other dementia. One SNP, rs2075650, located in an intron in TOMM40, had a genome-wide significant association with cognitive aging, which was replicated in the Swedish cohort. Fine SNP mapping of the TOMM40/APOE region revealed APOE (rs429358) and TOMM40 (rs11556505) loci associated with nonpathological cognitive aging. The high level of LD between APOE and TOMM40 makes it difficult to identify the associated SNP. Imputation and conditional analyses in the discovery and replication cohorts indicated that it may be APOE that is driving the observed association.
Another group, Caselli et al., found that both TOMM40 and APOE significantly influence age-related memory performance, but that they function independent of each other (Caselli et al., 2012). They speculated that if TOMM40 influences AAO in LOAD, either together with or instead of APOE, then it should have a similar effect on preclinical cognitive aging patterns. TOMM40 and APOE genotyping were performed on cognitively normal individuals 21–97 years of age. All study participants underwent longitudinal neuropsychological testing every 1–2 years until death. The onset of mild cognitive impairment or dementia, or the participant's decision to end participation was also recorded. Their results suggest that there is an early effect (before 60 years of age) attributable to the TOMM40 VL allele, but no significant APOE effect, and a separate late effect (after age 60) attributable to APOE ɛ4, characterized by accelerated memory decline, but no significant TOMM40 effect.
Similar findings were obtained by Johnson et al., (2011). They used mixed-effects regression models and compared healthy individuals (n = 117, mean age of 55 years) with an APOE ɛ3/ɛ3 genotype (with a neutral risk for AD) to individuals with VL/VL homozygous (n = 35), S/S homozygous (n = 38), and S/VL heterozygous genotypes (n = 44). They tested verbal memory measures and magnetic resonance imaging gray matter volumes in areas of the brain known to be affected in early AD. The VL/VL subgroup showed deficits on primacy retrieval from a verbal list learning task compared to the S/S group; this is also seen in early AD. A dose-dependent increase in the VL allele (from no VL alleles, to S/VL heterozygotes, to VL/VL homozygotes) was associated with a decrease in gray matter volume in the ventral posterior cingulate and medial ventral precuneus (a region of the brain affected early in LOAD). These findings indicate that individuals with VL alleles are at an increased risk of experiencing incipient LOAD-related cognitive and brain changes.
Diabetes mellitus (DM) is thought to be an established risk factor for dementia (Gudala et al., 2013) and studies have shown that diabetics exhibit reduced mean cortical thickness and reduced thickness in AD-vulnerable brain regions, in comparison to nondiabetics (Moran et al., 2015). Despite evidence linking AD-dementia to both TOMM40 and APOE, as well as to DM, prior studies have not directly examined whether these genotypes alter the association between blood glucose levels and structural brain measures. Wennberg et al. studied cognitively normal subjects (n = 233) and found (after adjustment for relevant factors, including age and medical conditions) that higher fasting blood glucose was significantly associated with decreased thickness in the parahippocampal gyrus and temporal pole, and reduced average thickness over AD-vulnerable regions (Wennberg et al., 2016). Interestingly, there was no evidence for greater cortical thinning in APOE ɛ4 carriers or in APOE ɛ3/3 individuals carrying the TOMM40 VL/VL genotypes. The associations between glucose levels and cortical thickness were no longer significant when individuals with glucose levels in the diabetic range were excluded from the analysis. These findings suggest that glucose levels in the diabetic range are associated with reduced cortical thickness in AD-vulnerable regions from as early as middle age. However, since they showed that neither TOMM40 nor APOE ɛ4 modified the relationship between blood glucose levels and cortical thickness, it suggests that these genotypes affect the risk for LOAD through independent mechanisms and not through the pathways in which DM and glucose metabolism alter the risk for AD dementia.
Deafness
Mohr-Tranebjærg syndrome (or X-linked deafness dystonia syndrome)
Mutations in the TIMM8A gene have been found to cause Mohr-Tranebjærg syndrome (MTS), an X-linked recessive progressive sensorineural hearing loss disease (Jin et al., 1996). TIMM8A, which resides in the IMS, belongs to a family of evolutionary conserved proteins and it is involved in mediating the import and insertion of hydrophobic membrane proteins into the IMM. Symptoms of MTS syndrome patients include prelingual or postlingual sensorineural hearing impairment, dystonia, ataxia and optic atrophy, and dementia and psychiatric symptoms (including personality changes and paranoia). It was first described in a Norwegian family with four generations of males affected with X-linked progressive deafness (Mohr and Mageröy, 1960).
Some of the mutations identified in TIMM8A include 151delT, 183del10, 108delG, and 127delT leading to loss-of-function frameshift mutations and a 21-kb deletion on Xq22, which resulted in the deletion of the entire gene (Tranebjaerg et al., 1995; Jin et al., 1996; Swerdlow and Wooten, 2001; Aguirre et al., 2006). TIMM8A stop mutations include a G to T transversion at position 105 (p.E24X), a C to T transversion at position 273 (p.R80X), and a C to T change at position 122 (p.Q38X) leading to the premature termination of the protein (Tranebjaerg et al., 2000, 2001; Ujike et al., 2001; Blesa et al., 2007).
In another study, it was found that a single base pair change (G to C at position 38 in exon 1) also resulted in the complete absence of the TIMM8A protein (Binder et al., 2003). This transversion resulted in the substitution of the methionine (M) start codon by an isoleucine (I) (p.M1I), which hindered protein translation. Another similar case was reported in 2015 where the start codon was eliminated by an A–G transversion, which possibly results in the absence of the TIMM8A protein (Penamora-Destriza et al., 2015). Interestingly, intronic mutations (IVS1–23A > C and IVS1 + 1G > T) resulting to RNA splicing defects were also identified in MTS patients (Ezquerra et al., 2005; Aguirre et al., 2008).
Notably, all mutations thus far have been predicted to result in a loss of function, truncation or absence of the protein except for a single missense mutation that is caused by a transversion from C to G at position 233 (p.C66W) (Tranebjaerg et al., 2000). TIMM8A forms a complex with one of the small TIMMs, TIMM13. The small TIMM proteins are kept in the IMS by either forming disulfide bonds or by binding to metal cofactors. It has been shown that TIMM8A-C66W leads to impaired zinc binding and it is suggested that this could cause the mutant protein to fold incorrectly and lose its ability to assemble with TIMM13 (Hofmann et al., 2002). Alternatively, it was suggested that C66W may prevent TIMM8A from interacting with CHCHD4 (MIA40, which promotes disulfide bond formation, as mentioned previously) and subsequently hinders disulfide bond formation (MacKenzie and Payne, 2007; Stojanovski et al., 2012).
Previous studies found that TIMM8A and TIMM13 form a 70 kDa complex in the IMS. This complex functions as a chaperone for IMS precursor proteins and is involved in the import of the IMM protein, TIMM23 (Roesch et al., 2002; MacKenzie and Payne, 2007). In the presence of TIMM8A-C66W, complex assembly is disrupted and TIMM23 import is decreased (Roesch et al., 2002). Therefore, impaired TIMM23 complex biogenesis could be the mechanisms underlying disease development since the TIMM23 complex is integral for the import of all matrix and many IMM proteins.
Meniere's disease
Meniere's disease (MD) is a disorder of the inner ear that is characterized by hearing loss, vertigo, and tinnitus. MD is a complex and pleiotropic disease where features such as neurodegeneration and loss of hair cells conflicted with previous study findings showing relatively normal cellular architecture (Ishiyama et al., 2015). Further research on the etiology of MD is necessary since most studies are performed on postmortem temporal bone and therefore, the exact cause of MD remains largely unknown. TOMM20 is widely expressed in the inner ear and cochlea of mice and humans, and a previous study found that the expression of TOMM20 was markedly decreased in the inner ear of aged individuals with MD (Balaker et al., 2013). Although this is the only study to implicate the decline in TOMM20 protein levels to MD, it could also be an indirect link due to the natural decline in TOMM20 levels with aging.
Cardiovascular diseases
Mitochondrial dynamics have become a key topic of interest in the field of cardiovascular disease (CVD) research; however, only a limited number of studies have investigated the involvement of mitochondrial protein import machineries, such as the TOMM and TIMM complexes, in these diseases:
TOMM20
Ischemic injury is damage that occurs in tissue when the blood supply is restricted, which then leads to a shortage of oxygen and glucose required for cellular metabolism. Ischemic preconditioning is a technique used to make tissue resistant to the loss of blood flow and oxygen. In 2006, Boengler et al. were the first to publish results from an in vivo model showing decreased TOMM20 expression after ischemic injury in isolated mitochondria from pig hearts (Boengler et al., 2006). They also showed the preservation of TOMM20 levels under ischemic preconditioning and that neither TOMM40 nor TIMM23 was affected by ischemic conditions, which could suggest a certain specificity for the changes observed for TOMM20 in ischemia/reperfusion.
TOMM22
TOMM22 and adenine nucleotide translocator (ANT) were recently identified as interacting partners of mitochondrial calcium-activated potassium (BKCa) cardiac channels (Zhang et al., 2017). This could suggest a functional role for the TOMM complex in cardiac mitochondria Ca2+ import and overall cellular survival. If TOM22 is defective or not present at the desired levels within the cell, the interaction with ANT could be disrupted, which could lead to inefficient cardiac cell functioning.
TOMM40
TOMM40 was previously found to be genetically associated with cardiovascular-related traits. A GWAS study, implementing a multivariate association test, was used to screen for SNPs associated with multiple biochemical traits related to cardiovascular risk, type 2 diabetes, or metabolic syndrome (Middelberg et al., 2011). This study screened 2548 adolescents and 9145 adults from 4986 Australian twin families. A subsequent univariate association analysis confirmed that two loci influenced more than one trait. The TOMM40/APOE-C1-C2-C4 gene cluster (rs2075650) was associated with low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, C-reactive protein, and triglycerides. Although no further studies found additional associations with cardiac-related traits, homozygous deletion of TOMM40 is lethal in mammals and hemizygous knockdown mice were shown to have cardiac phenotypes that deteriorated with age (Zeh, 2013).
TOMM70
A more recent study showed that TOMM70 protein was downregulated in hypertrophic hearts of animals and humans (Li et al., 2014). Upon further investigation, it was found that upregulation of TOMM70 protein levels could be cardioprotective under hypertrophic stress conditions. A recent study using mice cardiac tissue also found that TOMM70 levels were decreased in myocardial ischemia/reperfusion (MI/R) injury (Xue et al., 2017). These results showed that TOMM70 deficiency significantly worsened the MI/R injury and it negatively impacted on mitochondrial Ca2+ overload, which is thought to be the main cause of cardiomyocyte death. Mitochondrial Ca2+ uptake 1 (MICU1) is a molecule localized in the IMM, which regulates mitochondrial Ca2+ uptake and is downregulated by TOMM70 deletion. However, as previously reported, when TOMM70 levels were reestablished, the MI/R injury and mitochondrial Ca2+ were reduced and mitochondrial function was restored. This indicates that TOMM70, with the help of MICU1, serves an essential role in cardioprotection.
DNAJC19
Interestingly, a mitochondrial chaperone subunit of the TIMM complex was also previously implicated in CVDs in a syndrome called dilated cardiomyopathy with ataxia (DCMA; also called 3-methylglutaconic aciduria type V) (Ojala et al., 2012). The causative variant was a G to C substitution in the conserved AG splice site in intron 3 (IVS3–1 G > C) of the DNAJC19 gene (Davey et al., 2006). However, in a subsequent study, Ojala et al. found that two siblings with DCMA had a homozygous polymorphism (c.285A > C) (rs17850540) as well as a homozygous single-nucleotide deletion (c.300delA) leading to a premature termination of the protein 11 base pairs downstream (p.A100fsX11) (Ojala et al., 2012). This deletion results in DNAJC19 deficiency, which causes defects in the import of nuclear-encoded mitochondrial proteins, ultimately leading to the development of the disease. Thus, multiple mutations in this gene were shown to cause the same CVD.
Acylglycerol kinase
It has been shown that loss-of-function mutations in the AGK gene cause Sengers syndrome (Mayr et al., 2012). This syndrome, also known as cardiomyopathic mtDNA depletion syndrome 10, is an autosomal recessive mitochondrial disorder characterized by hypertrophic cardiomyopathy, congenital cataracts, skeletal myopathy, exercise intolerance, and lactic acidosis. AGK is a mitochondrial lipid kinase, but it was recently discovered that AGK is a subunit of the TIMM22 complex, where it plays a role in the import and assembly of mitochondrial carrier proteins in the inner membrane (Kang et al., 2017; Vukotic et al., 2017).
Cancer
Mitochondrial dysfunction is a common phenotype of cancer cells and is suspected to contribute to the development and progression of many different cancers. Differences between cancer cells and normal cells include variations in mitochondrial metabolic activity, mitochondrial molecular composition, and mtDNA variations, as well as changes of the nuclear genes encoding mitochondrial proteins (Aleskandarany et al., 2012). Since the TOMM and TIMM proteins are essential for mitochondrial function, many of these subunits have been associated with a variety of cancers:
TOMM34
Increased TOMM34 mRNA expression and its encoding protein is a reoccurring finding in colorectal cancer and has been shown to be involved in the growth of cancer cells (Shimokawa et al., 2006). It has also been found to be one of the top differentially expressed (upregulated) genes correlated with breast cancer metastasis and a marker of poor prognosis (Aleskandarany et al., 2012). The precise role of TOMM34 is still unclear; however, previous studies have shown that antibodies against TOMM34 inhibited translocation of preproteins into mitochondria, indicating that TOMM34 plays a role in mitochondrial preprotein import (Chewawiwat et al., 1999; Shimokawa et al., 2006; Aleskandarany et al., 2012). Recent studies also suggest that TOMM34 acts in a chaperone-like manner by maintaining an unfolded import-compatible configuration in newly synthesized precursors (Shimokawa et al., 2006; Aleskandarany et al., 2012).
TOMM20
High protein levels of TOMM20 were detected in anaplastic thyroid cancer compared to noncancerous thyroid tissue and is considered to be a negative prognostic marker when detected in gastric cancer patients (Curry et al., 2013; Johnson et al., 2015). Recent studies have also found high expression levels of TOMM20 protein in highly proliferative cancer cells of head and neck squamous cell carcinomas (Curry et al., 2013; Johnson et al., 2015). Evidence also suggests that TOMM20 is involved in cancer cell proliferation, but further studies are needed to determine the regulators of expression in cancer.
TIMM17
In humans, the TIMM17 gene is expressed as two isoforms, TIMM17A and TIMM17B. These isoforms are ubiquitously expressed in human tissue and assembled into the TIMM23 complex. TIMM17A and TIMM17B separate into two different TIMM23 complexes and only differ in their C-terminal structures (Bauer et al., 1999). Studies have shown that TIMM17A expression is increased in breast cancer and fibrolamellar carcinomas (Xu et al., 2010; Sokol et al., 2014). Xu et al. found that TIMM17A expression was absent in normal breast tissue, but could be detected during breast hyperplasia, which is an overgrowth of cells that line the milk glands inside the breast (Xu et al., 2010). Therefore, TIMM17A has been identified as a diagnostic marker and poor prognosis factor for breast cancer.
Interestingly, Rainbolt et al. (2013) showed that TIMM17A is a stress-regulated subunit of TIMM23. They found that pathologic insults (e.g., ER stress) result in decreased TIMM17A protein levels by reducing biogenesis and increasing degradation of the protein. This in turn decreases levels of TIMM23 complexes, which attenuates TIMM23-dependent protein import and facilitates induction of the mitochondrial unfolded protein response-associated genes. Therefore, TIMM17A plays an important protective role during cellular stress to prevent the accumulation of misfolded proteins within the mitochondria, which could lead to mitochondrial dysfunction.
CHCHD4
CHCHD4 was also found to have increased expression levels in tumor cells, specifically in pancreatic and breast cancer, and was shown to affect the levels of IMS proteins (Yang et al., 2012). When CHCHD4 was deleted, cells showed reduced tumor progression. In addition, another study suggested that the important tumor factor p53 is translocated to mitochondria in a CHCHD4-dependent manner and the downregulation of CHCHD4 resulted in the nuclear localization of p53 (Zhuang et al., 2016). This suppression of CHCHD4 was also shown to increase apoptotic response and decrease proliferation of cancer cells treated with 5-fluorouracil (a treatment commonly used in human cancer patients). Apart from this, it is interesting to note that two missense mutations in another family member of CHCHD4, namely CHCHD2, were associated with autosomal dominant PD (Funayama et al., 2015).
Therapeutic Applications and Interventions
This review highlighted the central role of mitochondrial protein import in the development of various diseases. Therefore, it is logical to conclude that these complexes should be investigated for their potential therapeutic applications.
A possible avenue for development of future therapies for AD involves an improved understanding of the life cycle of Aβ in mitochondria. This includes the mechanisms involved in its mitochondrial import by TOMM/TIMM machinery, its localization, as well as its proteolytic degradation. In this regard, drugs that result in decreased accumulation of this peptide are all viable options that warrant further investigation (Pinho et al., 2014). However, whether the pathology observed in AD can be altered by amending the levels of mitochondrial-associated Aβ is not currently known.
Also, α-synuclein, which has been implicated in PD and several other neurogenerative disorders, has been shown to be imported into mitochondria by the TOMM machinery. Future studies on the role of α-synuclein in mitochondria are needed and may prove to be important for the development of new therapeutic drugs for these conditions.
Immunotherapy is an active therapeutic approach designed to trigger the immune system to respond to tumor antigens and attack tumor cells (Miyata et al., 2015). Tumor-associated antigens (TAA) are proteins that are known to be overexpressed in malignant cells of various origins and are generally distributed. Using cDNA expression cloning methods these, TAA can be identified (Miyata et al., 2015). Antibodies against TOMM34 can possibly inhibit translocation of preproteins into the mitochondria and is therefore a promising target for the development of cancer peptide vaccines for colorectal cancer (Okuno et al., 2011, 2014; Aleskandarany et al., 2012; Hazama et al., 2014; Miyata et al., 2015). Clinical trials are underway, which show that peptide vaccines derived from TOMM34 and RNF43 (a protein that is associated with tumor cell proliferation), administered in combination with chemotherapy, are potentially beneficial in colorectal cancer (Okuno et al., 2011, 2014; Miyata et al., 2015).
Concluding Remarks
It is notable that very few pathogenic mutations have been identified in the TOMM/TIMM genes to date, attesting to the fact that these genes must be essential for life. This has been shown for TOMM40 where knocking out TOMM40 in Saccharomyces cerevisiae, Neurospora crassa, and mouse models was lethal (Baker et al., 1990; Taylor et al., 2003; Gottschalk et al., 2014). To date, TIMM8A and DNAJC19 are the only subunits in which multiple pathogenic/disease-causing mutations have been described (Table 1). Whether this is due to investigators mainly focusing on mutation screening in known disease-causing genes, rather than other possible TOMM/TIMM genes, or that there are just not many pathogenic mutations present in other complex subunits, remains unclear. Conversely, other subunits have been linked to human disease by their protein expression levels such as the increased expression of TOMM34 and CHCHD4 in cancer or the decreased expression of TOMM20 and TOMM70 in CVDs (Table 2).
Further investigations may reveal additional alterations at the level of mRNA and protein expression as a mechanism underlying various disorders. A possible area of interest could be post-transcriptional and post-translational modifications (PTMs). None of the studies reviewed investigated DNA methylation, phosphorylation, or any other form of PTMs as a cause of differential gene and/or protein expression. In addition, to expand our current knowledge on the involvement of TOMM/TIMM genes in human disease, the exploration of next-generation sequencing data should be considered. A myriad of studies is producing large amounts of publicly available data that are a potential goldmine for investigating genes/proteins such as the TOMM/TIMM complex subunits. Nevertheless, studies reviewed here found that these genes and proteins are implicated in disease development and these defects are associated with a variety of different diseases (Table 1 and 2). This emphasizes the essential, but understudied role of TOMM/TIMM genes in human health and disease.
These complexes also have the potential to be good candidates for therapeutic interventions, either through the upregulation/downregulation of various TOMM/TIMM complexes or through the blockage of the mitochondrial import of various deleterious molecules such as Aβ and toxic species of α-synuclein, which have been implicated in AD and PD, respectively. Further studies are needed to test this hypothesis and to determine the roles of these toxic molecules inside mitochondria.
It is important to note that proper protein expression, folding, transport, and clearance are critical for cellular function and overall health. Mitochondrial protein import is not a passive process, but a coordinated series of pathways requiring a number of components, recognition of targeting sequences, and the eventual insertion into the correct mitochondrial subcompartments, which is also dependent on proper PTMs. This implies that dysregulation at any stage of this process could result in dysfunctional mitochondria, ultimately impacting cellular survival. Although this review indicates that protein import machineries are implicated in a variety of disorders, these are relatively few. Notably, limited or no evidence was available for the involvement of MIA and SAM complexes in disease development. Further targeted studies may reveal that these complexes make an important contribution to human disease and show that their role to date has been understudied and underappreciated.
Footnotes
Acknowledgments
The authors would like to acknowledge Dr. Stefanie Malan-Muller and Prof. Helena Kuivaniemi for critical review of the article. The authors were supported by the National Research Foundation of South Africa (Grant no.: 106052) and the South African Medical Research Council (Self-Initiated Research Grant). A.N. was also supported by a Stellenbosch University Faculty of Medicine and Health Science Postdoctoral Fellowship. T.H. was supported by the PROMOS scholarship by the mobility program of the Deutscher Akademischer Austauschienst (DAAD) and the International Office of the Humboldt-Universität zu Berlin. We acknowledge the DST/NRF Centre of Excellence in Biomedical Tuberculosis Research, South African Medical Research Council Centre for Tuberculosis Research, Division of Molecular Biology and Human Genetics, and Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa.
Disclosure Statement
No competing financial interests exist.