
Abstract
The generation of reactive oxygen species (ROS) in response to oxidative stress has important effects on cell development, normal function, and survival. It may cause oxidative damage to intracellular macromolecular substances and mitochondria through several signaling pathways. However, the damaged mitochondria promote further ROS generation, creating a vicious cycle that can cause cellular injury. In addition, excessive ROS produced by damaged mitochondria can trigger mitophagy, a process that can scavenge impaired mitochondria and reduce ROS level to maintain stable mitochondrial function in cells. Therefore, mitophagy heaps maintain cellular homeostasis under oxidative stress. In this article, we review recent advances in cellular damage caused by excessive ROS, the mechanism of mitophagy, and the close relationship between ROS and mitophagy. This review provides a new perspective on therapeutic strategies for related diseases.
Introduction
M
Cells are equipped with enzymatic and nonenzymatic antioxidants such as glutathione (GSH), superoxide dismutase (SOD), and catalase (CAT), to avoid oxidative stress. However, the antioxidant capability of cells becomes insufficient under stressful environmental conditions, resulting in excessive ROS production (Schieber and Chandel, 2014; Li et al., 2015). ROS overproduction can cause mitochondrial swelling and opening of mitochondrial permeability transition pores (MPTP), consequently promoting the release of apoptosis factors such as cytochrome C and finally inducing apoptosis (Scherz-Shouval and Elazar, 2011). Meanwhile, ROS could trigger the mitochondria to generate more ROS, constituting a positive feedback cycle (Schieber and Chandel, 2014). Therefore, the maintenance of ROS production or accumulation is crucial for normal cellular homeostasis.
In consideration that dysfunctional or damaged mitochondria are risk factors to cell survival, timely removal of these impaired mitochondria is greatly important for maintaining their quality and quantity and cell homeostasis (Ashrafi and Schwarz, 2013). ROS generated by damaged mitochondria can trigger mitophagy, which can selectively scavenge dysfunctional or damaged mitochondria and reduce ROS level to ensure stable mitochondrial function in cells and ultimately maintain cell survival (Youle and Narendra, 2011; MacVicar, 2013). Therefore, rational regulation of mitophagy effectively improves cellular resistance to oxidative stress and reduce or delay injury. However, underlying mechanism of the interaction between ROS and mitophagy is still poorly understood. This article summarizes and discusses the cellular damage caused by excessive ROS, the mechanism of mitophagy, and the close relationship between ROS and mitophagy.
Excessive ROS Inducing Cell Insults
Brief overview of ROS
ROS are inevitable by-products of mitochondrial metabolism (Murphy, 2009). Oxidative stress is due to ROS overproduction or inadequate ROS removal, and large amounts of ROS could injure the organelles, cells, or even bodies. Mitochondria are not merely organelles that produce energy and regulate cellular signaling transduction and apoptosis but also the main source of ROS generation (Murphy, 2009). ROS are composed of polar molecules, including O2 −, OH−, and HO2 −, and nonpolar molecules, such as H2O2. In addition, ROS are produced by the mitochondrial electron transport chain and NADPH oxidase (NOX) (Schieber and Chandel, 2014; Li et al., 2015). However, NOX, which actively produces superoxide across the membranes of neutrophils and phagosomes, contributes to bacterial autophagy action (Chen et al., 2007). Many antioxidant systems, including SOD, CAT, and redox factor 1, eliminate ROS overproduction in cells, and they could protect cells from oxidative stress-induced attack.
Major signaling pathways of cell injury induced by ROS
Excessive ROS accumulation is often associated with an imbalance between oxidative and antioxidative systems, resulting in damaged lipids, proteins, and DNA (Cross et al., 1987). In addition, ROS overproduction leads to the opening of MPTP, a state that allows the mitochondrial outer membrane (MOM) to be permeable and promotes the leakage of apoptosis-amplification factors such as cytochrome C and apoptosis inducing factor, resulting in apoptosis (Pervaiz et al., 2009). ROS overproduction leads to stress-induced premature senescence that is similar to replicative senescence, in terms of exhibiting morphological changes with flattening and enlargement and upregulation of SA-β-gal activity (Yang et al., 2015). At present, ROS-inducing injury involves the following three aspects (Fig. 1).
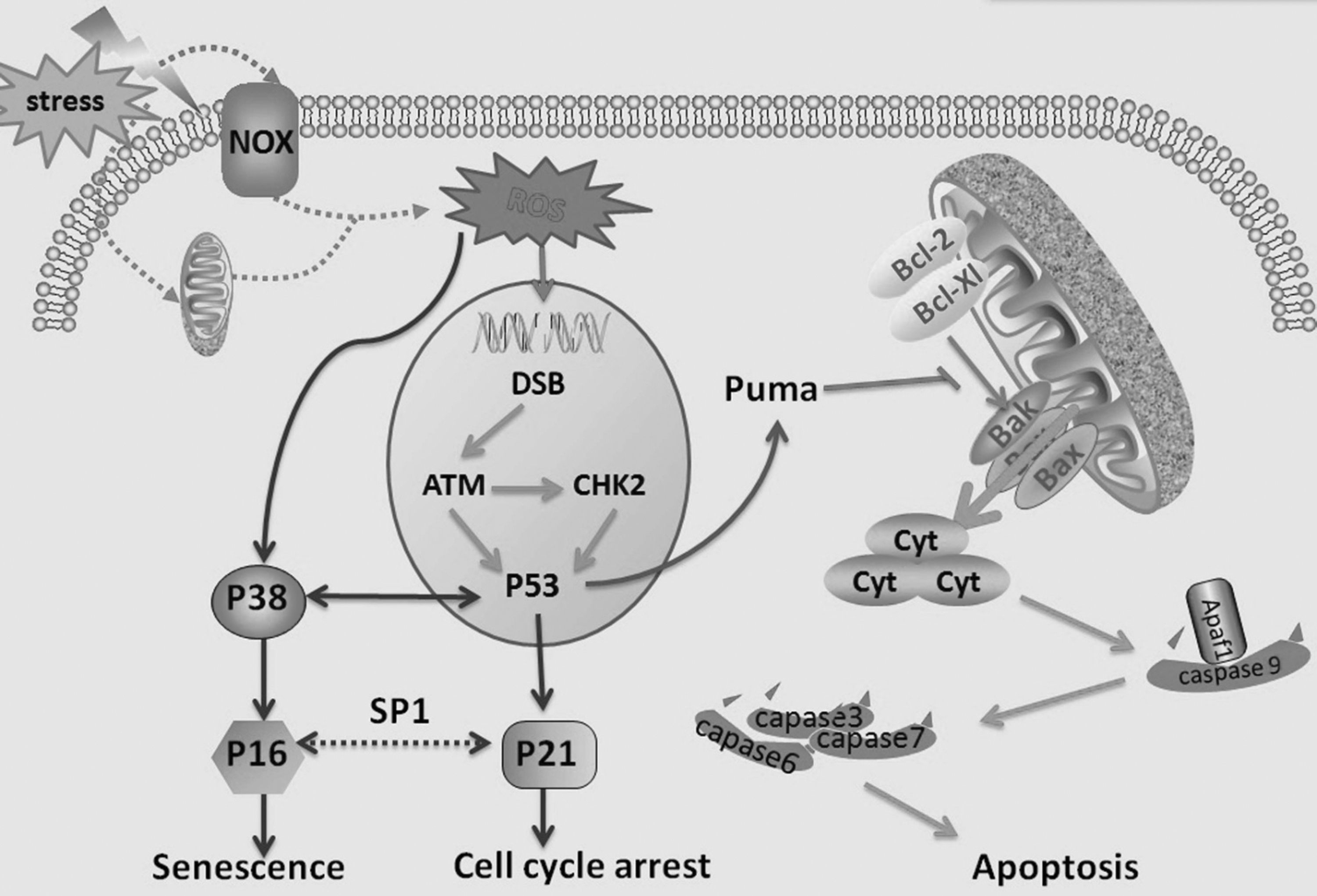
Molecular signal pathways of ROS-induced cell injury. DSBs induced by ROS activate ATM kinase, which could phosphorylate CHK2 and p53. The phosphorylated p53 may act upstream of p21 and p16 to cause cell cycle arrest and senescence, respectively. Furthermore, ROS can facilitate the expression of p38 that may induce cellular senescence by activating p16 or interacting with p53. Meanwhile, Puma is a downstream target of p53 and could block the interaction of antiapoptotic proteins, such as Bcl-2 and Bcl-xl, with proapoptotic factors, such as Bax and Bak, leading to the interruption of mitochondrial integrity. Subsequently, caspase-activating factors such as cytochrome C and Apaf-1 are released from the mitochondria, initiate caspase-9, and then set off the signaling cascades (caspases-3, -6, and -7), resulting in apoptosis. ATM, ataxia telangiectasia mutated; CHK2, checkpoint kinase 2; DSB, double-strand break; ROS, reactive oxygen species.
DNA damage response pathway
Among various cellular macromolecules, DNA is the most crucial target for ROS-induced cell senescence and apoptosis (Kim et al., 2016a). These DNA lesions have many types, among which double-strand breaks (DSBs) are the most detrimental. Importantly, DSBs could activate numerous metabolism reactions, particularly DNA damage response (DDR) to repair DSBs or remove the insulted cells through the action of senescence or apoptosis to maintain genome integrity. Furthermore, this process should be mediated by ataxia telangiectasia mutated kinase, which could phosphorylate p53 and checkpoint kinase 2. In addition, phosphorylated p53 upregulates the production of p21 and Puma to induce cell cycle arrest and apoptosis, respectively (Shao et al., 2014; Nair et al., 2015; Yang et al., 2015).
NF-κB pathway
NF-κB is a nuclear transcription factor that is sensitive to ROS accumulation, and it consists of five members: p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1), and p52/p100 (NF-κB2). These members often dimerize with each other, except RelB. In addition, the heterodimers indwell in cytoplasm in association with IκBs, which suppress their nuclear translocation and phosphorylation (Hui et al., 2014). In general, ROS can activate IKKβ and phosphorylate IκBα. Furthermore, the NF-κB heterodimers undergo nuclear translocation, as well as bind to DNA κB site, which triggers the p53/p21 axis to accelerate premature senescence after ubiquitination and degradation of phosphorylated IκBα (Bhattacharyya et al., 2009; Sfikas et al., 2012; Lee et al., 2014).
p38 mitogen-activated protein kinase pathway
p38 is a major member of the family of mitogen-activated protein kinases (MAPKs) involved in the regulation of cell cycle, senescence, and apoptosis (Xu et al., 2014; Zhou et al., 2015). Its signaling cascade involves sequential activation of MAP kinase kinases (MAPKKs), including MEKK3 and MEKK6. These kinases are initiated by one of the several upstream MAPKKs, such as apoptosis signal-regulating kinase, dual-leucine zipper-bearing kinase 1. A plethora of evidence indicates that ROS oxidizes Trx from ASK-1 for activation, leading to the activation of P38 pathway (Katagiri et al., 2010; Shao et al., 2011). In addition, ROS-activated p38 pathway plays a pivotal role in the induction of premature senescence of stem cells through upregulating of p16 and/or interacting with p53 (Ito et al., 2006; Kim et al., 2008; Shao et al., 2011).
p53/p21 and p16-Rb pathways
The mentioned findings all involve the p53/p21 and p16-Rb pathways. Phosphorylation of p53 can induce the expression of p21, proapoptotic proteins such as Puma, and p16 to trigger cell cycle arrest, apoptosis, and senescence, respectively. In addition, Puma can inhibit the interactions of proapoptotic effectors (e.g., Bax and Bak) with the antiapoptotic proteins (e.g., Bcl-xl and Bcl-2). This phenomenon leads to the interruption of mitochondrial integrity and the release of caspase-activating factors such as cytochrome C and Apaf-1, which could initiate caspase-9 and then the effector caspases, ultimately resulting in stem cell self-destruction by apoptosis (Shao et al., 2011, 2014; Insinga et al., 2014). The N-terminus of p16 has a structure homologous to cyclin D, which competes with cyclin D for binding to cyclin-dependent kinase 4/6. This phenomenon causes Rb hypophosphorylation and inhibits the initiation of E2F transcription factor, thereby restricting of G1/S cell cycle progression and senescence (Sharpless and DePinho, 1999; Narita et al., 2003; Yue et al., 2014). In addition, p16 can stabilize p21 protein, which, in turn, activates the expression of the p16 gene through Sp1 transcription factor and synergistically restricts cell cycle (Gu et al., 2013).
Mechanism of Mitophagy
From the mentioned research, we can conclude that excessive ROS produced by damaged mitochondria could threat cell health. Therefore, the clearance of damaged or aging mitochondria through autophagy, a process called mitophagy, is vital for cells to maintain their proper mitochondrial quality. Previous studies using the autophagosome marker MAP1 light chain 3 (LC3: mammalian autophagosomal homolog of yeast Atg8) described mitophagy as the process of engulfment of damaged or aging mitochondria into isolation membranes (MacVicar, 2013). Then, the newly formed vesicles seal and fuse with lysosomes to clear the engulfed mitochondria (Kim et al., 2007). Currently, mitophagy receptors, such as Atg32 in yeast and BNIP3, NIX, and FUN14 domain containing 1 (FUNDC1) in mammals, play an important role in removing of mitochondria (Okamoto et al., 2009; Kanki, 2010). The PTEN-induced putative kinase 1 (Pink1)-Parkin signaling pathway can also induce mitophagy through an ubiquitylation-related process if mitochondrial membrane potential is reduced (Wu and Chen, 2015; Xiao et al., 2017).
Mitophagy in yeast
Genome-wide screening showed that Atg32 is a mitochondrial receptor for mitophagy in yeast. It is an MOM protein with N- and C-terminal domains exposed to cytosol and mitochondrial intermembrane space, respectively (Okamoto et al., 2009; Kondo-Okamoto et al., 2012). Its cytosolic domain contains a tetrapeptide sequence WXXI region, which functions as the Atg8-family interacting motif (AIM, equivalent to LC3-interacting region [LIR]) and is important for the interaction of Atg32 with the isolation membrane protein Atg8 (Okamoto et al., 2009). Kondo-Okamoto et al. (2012) reported that Atg32 with AIM-deficient mutant exhibits only partial but not absolutely deficient mitophagy, but it cannot interact with Atg8, suggesting that AIM-dependent Atg32-tg8 binding is important but not essential for mitophagy. In addition, Atg32 can bind with Atg11, which is a selective autophagy receptor that can recruit a range of cargos into autophagosomes through interacting with Atg8 (Kondo-Okamoto et al., 2012; Levchenko et al., 2016). Under nitrogen starvation, Atg32 phosphorylation is remarkably increased, which promotes Atg32–Atg11 interaction and subsequent mitophagy. Activation of Hog1, a MAPK, relies on its phosphorylation by Pbs2 that is an MAPKK. A recent study has revealed that casein kinase-2 (CK-2), initiated by Hog1, can phosphorylate Atg32 at Ser114 and Ser119, increasing the stability of Atg32–Atg11 interaction and promoting mitophagy (Aoki et al., 2011; Kanki et al., 2013; Liu et al., 2014). However, the mechanism by which Hog1 promotes the phosphorylation of Atg32 by CK-2 needs to be investigated (Fig. 2A).
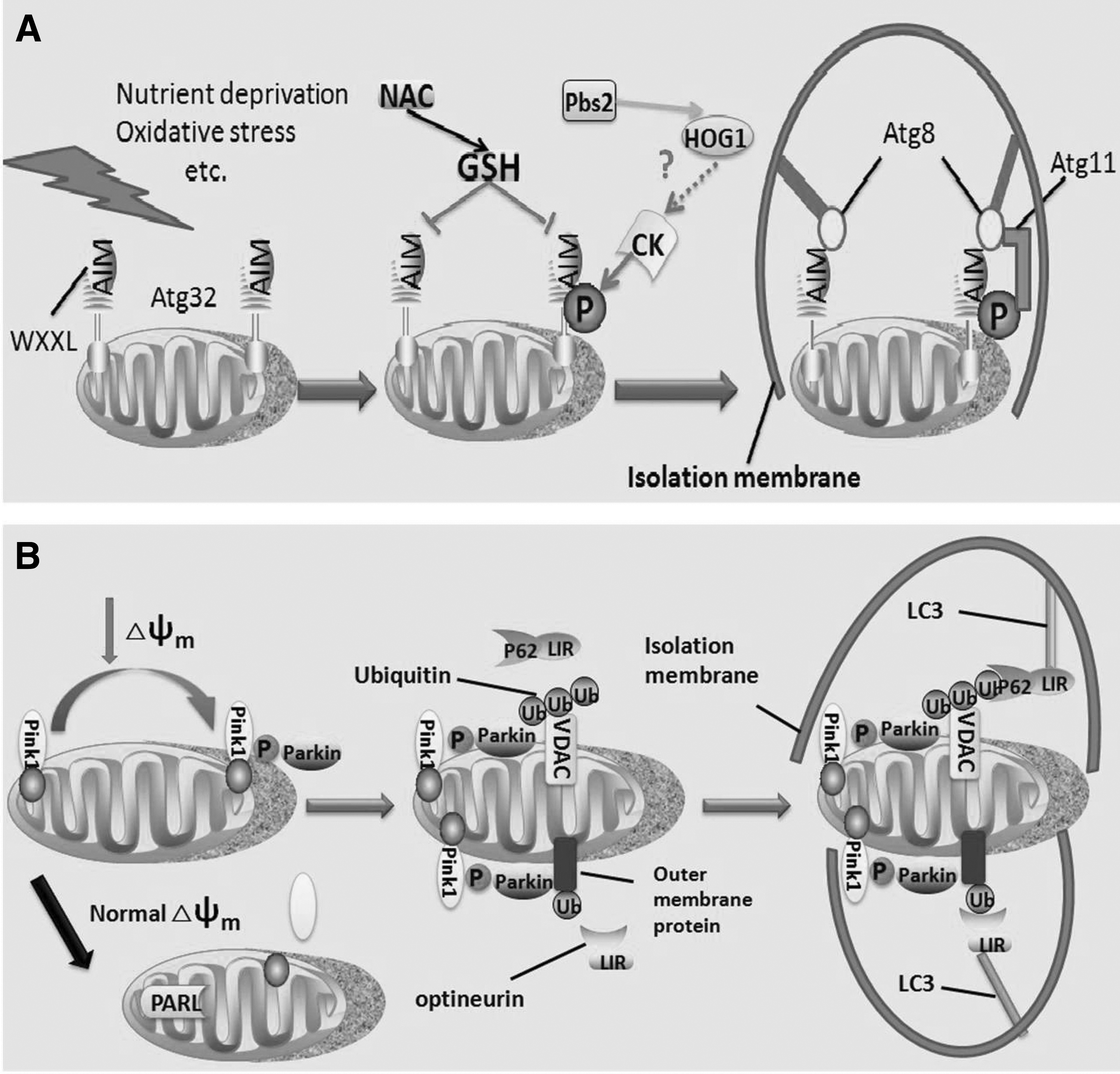
Moreover, N-acetylcysteine (NAC), an antioxidant that can scavenge free radicals such as ROS, could inhibit Atg32 expression and consequent mitophagy (Kondo-Okamoto et al., 2012). Interestingly, Deffieu et al. (2009) reported that NAC can suppress mitophagy through increasing the cellular GSH pool rather than removing ROS. However, the exact role of ROS in Atg32-mediated mitophagy needs to be clarified.
Mitophagy in mammalian cells
Pink1–Parkin signaling pathway
Pink1, a nuclear-code mitochondrial kinase, and Parkin, a cytosolic E3 ubiquitin ligase, regulate mitophagy (Xiao et al., 2017). Pink1 and Parkin work through identical pathways, with Pink1 acting upstream of Parkin (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). The molecular mechanism by which Pink1 and Parkin act together to mediate mitophagy depends on the mitochondrial membrane potential. Pink1 is expressed and transported into all mitochondria and then rapidly cleaved and degraded by presenilin-associated rhomboid-like protease (PARL) at the inner membrane of healthy mitochondria (Greene et al., 2012; Wu and Chen, 2015; Checler et al., 2017). Nevertheless, upon mitochondrial damage and dissipation of membrane potential, Pink1 is stabilized on the MOM and recruits Parkin to impaired mitochondria (Matsuda et al., 2010; Okatsu et al., 2012, 2015). However, the manner of regulating Pink1 cleavage and degradation by membrane potential remains poorly understood. Furthermore, mir-27a/b could inhibit Pink1 accumulation, and PKA activity controls Pink1 stability upon impaired mitochondria (Akabane et al., 2016; Kim et al., 2016b).
Pink1 accumulation and activation on the MOM promote the recruitment of cytosolic Parkin on the depolarized mitochondria. Pink1 may phosphorylate ubiquitin at Ser65, and the phosphorylated ubiquitin activates Parkin, which is essential for Parkin translocation and mitophagy (Kane et al., 2014; Akabane et al., 2016; Xiao et al., 2017). Once activated, Parkin ubiquitinates diverse MOM proteins in depolarized mitochondria, such as voltage-dependent anion channels (VDACs) and Mfn1/2 (Sun et al., 2012; Norris et al., 2015). Furthermore, these proteins may mediate the subsequent sequestration of mitochongdrial transport to the isolation membrane through several adaptor proteins (e.g., P62 and optineurin), which can directly interact with LC3 (Geisler et al., 2010; Kane et al., 2014).
After VDAC recruitment on the mitochondrial membrane, the Lys27 poly-ubiquitin chains are formatted on VDACs by Parkin. P62 can directly interact with ubiquitin chains through its C-terminal ubiquitin-binding domain and with LC3 through LIR motif. Although p62 plays an important role in the linkage of aggregated proteins with LC3 and subsequent mitophagy, the essential molecule for this process remains controversial (Komatsu et al., 2007; Narendra et al., 2010; Okatsu et al., 2010). Optineurin is another autophagy receptor that possesses an ubiquitin-binding domain and LIR motif (Wong and Holzbaur, 2014). Once associated with ubiquitinated MOM proteins, optineurin recruited on impaired mitochondria in a Parkin-dependent manner. Subsequently, it induces mitophagy through its interaction with LC3 by LIR without p62 (Wong and Holzbaur, 2014, 2015). In particular, Parkin ubiquitinates mitofusins, which might facilitate their degradation and then promote mitochondrial fragmentation, resulting in mitophagy (Gegg and Schapira, 2011; Ding et al., 2012). Furthermore, Parkin inhibits the refusion of impaired mitochondria with healthy mitochondria and ultimately segregates damaged mitochondria for mitophagy through the degradation of mitofusins that facilitate mitochondrial fusion (Tanaka et al., 2010; Gegg and Schapira, 2011; Ding et al., 2012) (Fig. 2B).
NIX and BNIP3 pathway
BNIP3 (B cell lymphoma2 [BCL-2]/adenovirus E1B 19KDa interacting protein 3) and NIX (also known as BNIP3L) were initially identified as BCL-2 homology 3 domain (BH3)-only proapoptotic proteins (Boyd et al., 1994; Ohi et al., 1999). Both of them contain a carboxyl-terminal transmembrane domain through which they are inserted into the MOM, whereas their N-terminal domain faces the cytoplasm (Imazu et al., 1999; Ohi et al., 1999). As alternative BH3-only proteins, BNIP3 and NIX, confer similar proapoptotic activity, which alters the permeability of mitochondrial membrane and increases the release of cytochrome C through heterodimerizing with BCL-2 or B cell lymphoma extra large (BCL-XL) (Imazu et al., 1999). During the red blood cell differentiation and maturation, NIX is highly expressed and is involved in eliminating of mitochondria from reticulocytes (Novak et al., 2010). It contains a typical LIR motif that can interact with LC3 protein and its homolog GABA receptor-associated protein (GABARAP), thereby directly recruiting isolation membranes to mitochondria (Ding et al., 2010b; Novak et al., 2010). BNIP3 also contains an LIR motif, which is phosphorylated at Ser17 and Ser24 and facilitates its interaction with LC3 (but not GABARAP) to induce mitophagy (Zhu et al., 2013). However, the mechanism by which NIX as an autophagy receptor targets damaged mitochondria and induces mitophagy remains unknown.
Treatment with carbonyl cyanide p-trifluoromethoxyphenylhydrazone could induce mitochondrial depolarization and rescue mitophagy in NIX−/− erythroid cells, suggesting that NIX promotes mitophagy, possibly due to its role in inducing mitochondrial depolarization (Sandoval et al., 2008). Ding et al. have reported that NIX can induce mitochondrial depolarization that may initiate parkin ubiquitin-p62-mediated mitochondrial priming by regulating the recruitment of Pink1 or suppressing Pink1 kinase proteolytic cleavage (Ding et al., 2010b; Zhang et al., 2016). However, the exact mechanism by which NIX regulates mitochondrial membrane potential is still controversial. BNIP3 may induce mitochondrial depolarization in murine embryonic fibroblasts through BAX or BAK, resulting in the opening of MPTP (Kubli et al., 2007). In addition, BCL-XL can inhibit the mitochondrial membrane potential dissipation induced by NIX (Imazu et al., 1999). Nevertheless, in reticulocytes, NIX-mediated mitophagy does not depend on BAX and BAK, and NIX deficiency has no distinct impact on mitochondrial depolarization (McLelland et al., 2014). Furthermore, Parkin can ubiquitinate NIX, thereby facilitating it to be recognized by the autophagosome receptor (e.g., LC3), subsequently inducing mitophagy (Gao et al., 2015) (Fig. 3A). As a BH3-only protein, whether NIX or BNIP3 can compete with Beclin-1 for binding to BCL-2 or BCL-XL and then induce mitophagy needs to be further studied.
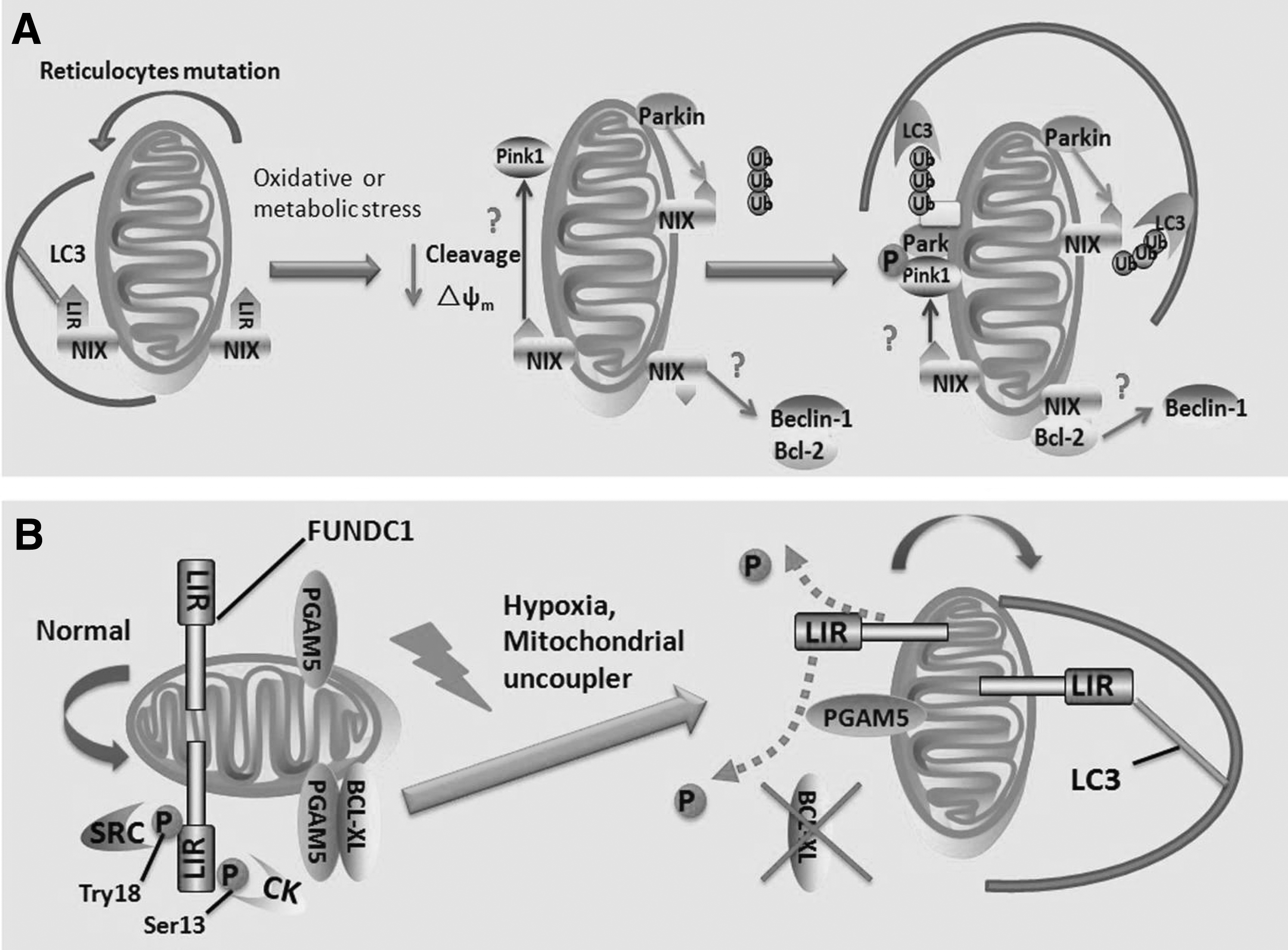
FUNDC1 pathway
FUNDC1, an MOM protein, is involved in mitophagy in mammalian cells (Liu et al., 2012). It functions as a mitophagy receptor that contains three transmembrane domains, including the N- and C-terminal regions and the intermembrane space. FUNDC1 has an LIR motif in its N-terminal that interacts with LC3, similar to yeast Atg32 and NIX (Liu et al., 2012). Under normal conditions, mitophagy induced by FUNDC1 is inhibited through its phosphorylation by Src kinase or CK-2 at Tyr18 in the LIR motif and Ser13, respectively. These phosphorylations inhibit the FUNDC1–LC3 interaction (Liu et al., 2012; Chen et al., 2014). During hypoxia or mitochondrial uncoupler treatment, Src and CK-2 are inactivated, and the degradation of Bcl-xl can release PGAM5, a mitochondrial phosphatase, and then dephosphorylates FUNDC1 at Ser13. This dephosphorylation facilitates FUNDC1–LC3 interaction, initiating the sequestration of mitochondria through the isolation membrane (Liu et al., 2012; Lu et al., 2014; Wu et al., 2014) (Fig. 3B).
Initiation of Mitophagy by ROS
As already described, pathological conditions, such as hypoxia, nutrient starvation, ischemia/reperfusion (IR), or metabolic stress, can stimulate cellular ROS production, leading to oxidative damage that induces mitochondrial dysfunction and cell injury (Chakrabarti and Jahandideh, 2014; Fandy et al., 2014). Meanwhile, the relative excess accumulation of ROS involved mitophagy, which can reduce oxidative damage and ROS production. The regulation mechanism of mitophagy by ROS includes the ROS-Pink1/Parkin-mitophagy, ROS-hypoxia-inducible factor-1 (HIF)-Bnip3/Nix-mitophagy, ROS-FOXO3-LC3/BNIP3-mitophagy, and ROS-NRF2-P62-mitophagy signaling pathways (Tracy et al., 2007; Zhao et al., 2007; Aucello et al., 2009; Taguchi et al., 2011; Wang et al., 2012; Jaramillo and Zhang, 2013; Wei et al., 2014; Shen et al., 2015; Klumpen et al., 2017) (Fig. 4).

Regulation of mitophagy by ROS. ROS are mainly produced in mitochondria. Under certain conditions such as hypoxia, starvation, IR, and metabolic stress, ROS accumulate and induce oxidative damage in cells. In nucleus, excessive ROS activate HIF-1, FOXO3, and NRF2, which stimulate the transcription of BNIP3/NIX, LC3/BNIP3, and P62, respectively, consequently facilitating mitophagy. Furthermore, an acute burst of ROS may reduce membrane potential and subsequently initiate the PINK1/Parkin signaling pathway to promote mitophagy. HIF-1, hypoxia-inducible factor-1; IR, ischemia/reperfusion.
ROS-Pink1/Parkin-mitophagy
As mentioned before, the Pink1/Parkin signaling pathway initiates mitophagy. Furthermore, an acute burst of ROS produced by damaged mitochondria can reduce mitochondrial membrane potential. The depolarized mitochondria stabilize Pink1 and subsequently recruit Parkin to the MOM that ubiquitinates several MOM proteins. Thereafter, the injured mitochondria are engulfed by LC3 and finally transported to lysosomes (Wang et al., 2012; Wei et al., 2014). Meanwhile, Parkin can cause the degradation of mitofusions (MFN1 and MFN2), which are proteins necessary for the fusion of mitochondrial membranes, and subsequently promote mitochondrial fragmentation that facilitates the quarantine of damaged mitochondria and their autophagosomal engulfment (Poole et al., 2010; Wang et al., 2012; Rojas-Charry et al., 2014).
ROS-HIF-BNIP3/Nix-mitophagy
HIF-1, a crucial factor in the cells' response to hypoxia conditions, could promote the transcription of BNIP3 and NIX (Tracy et al., 2007; Mahalingaiah and Singh, 2014). As mentioned before, these proteins could directly or indirectly initiate Pink1/Parkin signaling pathway, resulting in mitophagy (Ding et al., 2010b; Zhang et al., 2016). Furthermore, they could also compete with Beclin-1 to bind with BCL-2 or BCL-XL, thereby leading to mitophagy (Zhang et al., 2008).
Nevertheless, ROS produced by damaged mitochondria activate HIF-1 (Klumpen et al., 2017). On the one hand, under hypoxic condition, ROS could inhibit the activity of prolyl hydroxylase that promotes HIF-1 degradation through E3 ubiquitin ligases, subsequently stabilizing HIF-1 (Li et al., 2014). On the other hand, a modest increase in ROS stabilizes Sentrin/SUMO-specific proteases (SENPs) through inhibiting its degradation by the ubiquitin–proteasome pathway. In addition, ROS regulate SENP3 to redistribute from the nucleolus to the nucleoplasm. The stabilization and redistribution of SENP3 promote the transcriptional activity of HIF-1 (Huang et al., 2009).
Therefore, ROS could induce the expression of HIF-1 and consequently facilitate mitophagy by initiating downstream target genes, such as BNIP3 and NIX.
ROS-FOXO3-LC3/BNIP3-mitophagy
Aucello et al. found that increasing levels of ROS may initiate FOXO3 signaling pathways in muscle cells and subsequently trigger both the ubiquitin–proteasome pathway and transcription of several Atgs, such as BNIP3 and LC3 (Mammucari et al., 2007; Aucello et al., 2009; Shen et al., 2015); these protein products could induce the formation of mitophagy in response to oxidative damage. Moreover, the transcription of several other mitophagy-related genes such as Beclin1, GABARAP, and ULK2, may be regulated by FOXO3 during starvation (Zhao et al., 2007).
ROS-NFR2-P62-mitophagy
As already described, p62 contributes to the selective autophagy of depolarized mitochondria because it contains an LIR motif, which interacts with LC3. The interaction plays an important role in mitophagy, including isolation membrane fusion, cargo selection, and autophagosome transport.
NRF2, a transcription factor of the basic leucine zipper family, is triggered by oxidative stress and subsequently binds to the antioxidant-responsive element located in the p62 promoter to incite its mRNA expression (Jain et al., 2010; Rubio et al., 2014). Under normal condition, NRF2 is constitutively degraded by its interaction with the E3 ubiquitin ligase KEAP1 (Kelch-like ECH-associated protein 1) (Taguchi et al., 2011; Jaramillo and Zhang, 2013). Moreover, p62 could promote the transcription of NRF2 because it possesses a keap1-interacting region that can release NRF2 from KEAP (Puissant et al., 2012). Hence, in this procedure, p62 could activate NRF2 and then drive its own transcription, leading to a positive feedback cycle (Jain et al., 2010). However, some controversies about the details of the interaction between P62 and KEAP1 still remain to be solved.
Related Diseases and New Perspectives on Therapeutic Strategies
As mentioned before, mitophagy and ROS play a pivotal role in mitochondrial homeostasis and quality control. Experimental evidence indicates that the accumulation of damaged mitochondria has been associated with various human diseases, such as neurodegeneration, cancer, and metabolic disorders. These diseases display characteristic defective energy metabolism because of the accumulation of dysfunctional mitochondria and ROS.
Releated diseases
Neurodegenerative diseases
The accumulation of ROS and dysregulation of mitophagy have been involved in several types of neurodegenerative diseases, such as Parkinson's disease (PD), Alzheimer's disease (AD), and Huntingtong's disease (HD) (Cha et al., 2015; Nah et al., 2015).
PD is caused by insufficient dopamine production in substantia nigra, and the accumulation of ubiquitinated α-synuclein-containing inclusions within dopaminergic neurons, called Lewy bodies (Nah et al., 2015). Several lines of evidence have been reported that Pink1/Parkin-mediated mitophagy and damaged mitochondria caused by extensive ROS have revealed close relationship with PD, consistent with the accumulation of dysfunctional mitochondria in PD patients (Dubinsky, 2005; Geisler et al., 2010; Jiang and Mizushima, 2014). The mutation of Pink1 or Parkin and extensive ROS lead to both the accumulation of dysfunctional mitochondria and degeneration of dopaminergic neurons (Park et al., 2006, 2018).
Besides, defective mitochondria and dysregulation of mitophagy are strongly associated with AD (Du et al., 2010). AD is characterized by progressive dementia and degeneration and eventual loss of brain neurons, containing aberrant extracellular deposition of β-amyloid protein (Aβ) (Wang et al., 2017). Specifically, extensive Aβ generation in mitochondria causes mitochondrial dysfunction, inducing oxidative stress. This oxidative stress aggravates mitochondrial damage and then promotes AD progression (Casley et al., 2002). It was reported that Parkin expression can decrease Aβ levels and clear dysfunctional mitochondria and reduce oxidative stress through mitophagy, maintaining neuron homeostasis (Khandelwal et al., 2011; Wang et al., 2014).
Moreover, defective mitochondria, such as decreased mitochondrial membrane potential, reduced mitochondrial movement, and aberrant mitochondrial ultrastructures have been found in HD patients and mouse models (Miwa et al., 2003; Martinez-Vicente et al., 2010). In addition, neuron cells isolated from HD have abnormal mitochondria turnover rate and defective mitophagy. Therefore, it is proposed that mitophagy plays a protective role in the HD process.
Cancer
Dysfunctional mitochondria and accumulation of ROS have been associated with cancer progression, consistent with the idea that Parkin is crucial for mitophagy and tumor suppression (Chourasia et al., 2015a; Kulikov et al., 2017). Parkin regulates several significant cancer cell biology processes, such as mitochondrial homeostasis, stress resistance, and cell cycle. Recent studies have suggested that Parkin mutations have correlated with colon, breast, and lung cancer, and revealed that Parkin deletion promoted liver cancer progression (Zhang et al., 2011; Shah et al., 2012; Rao et al., 2014; Bernardini et al., 2017). These reports further suggested that Parkin may suppress tumorigenesis.
Other important mitophagy regulators, BNIP3 and Nix, also suppress cancer development (Bernardini et al., 2017). BNIP3 is suppressed in liver, colorectal, and pancreatic cancer, and this loss leads to mitophagy dysregulation, increases ROS production, and accumulation of damaged mitochondria, facilitating progression of pancreatic cancer and promoting metastasis of breast cancer cells in mouse models (Murai et al., 2005; Chourasia and Macleod, 2015; Chourasia et al., 2015b). Interestingly, the increase in ROS caused by defective mitophagy contributes to tumorigenesis and metastasis through inducing mitochondria damage (Prasad et al., 2017).
Alternatively, evidence indicates that mitophagy may promote tumorigenesis by enhancing cellular adaptation to stress during the late stage of tumor growth (Bernardini et al., 2017; Yan et al., 2017).
Cardiovascular and hepatic disease
Mouse models with deficient mitophagy are prone to having various cardiac diseases, these illustrate that mitophagy plays an important role in cardiac protection (Suarez-Rivero et al., 2016; Nan et al., 2017). Cardiac ischemia reduces cellular ATP content, which leads to energy stress and abnormal ROS production associated with mitochondrial dysfunction (Lesnefsky et al., 2001). Mitophagy triggered by ROS protects against these injuries during IR by eliminating intracellular waste material and dysfunction mitochondria that cause abnormal ROS generation.
PINK1 and Parkin mutations increase accumulation of dysfunctional mitochondria and ROS, and aggravate IR injury. In heart ischemic preconditioning mouse models, Parkin-mediated mitophagy alleviates IR injury (Huang et al., 2011). Similarly, upregulation of BNIP3 promotes mitophagy and resists various apoptotic factors, which further alleviate IR injury (Ma et al., 2015). Hoshino et al. (2013) found that cytosolic p53, mitophagy-negative regulatory factor, facilitates mitochondrial dysfunction and heart failure in mice models. Interestingly, exaggerated mitophagy activation during excessive ROS production may trigger cell death, and insufficient mitophagy negatively impacts cardiac remodeling because of accumulation of damaged mitochondria and misfolded proteins (Matsui et al., 2007; Zhai et al., 2011).
Moreover, dysfunctional mitochondria and dysregulation of mitophagy have close relationship with hepatic diseases. It has been reported that overdose of acetaminophen could cause hepatic necrosis through damaging mitochondria, and mitophagy plays a protective role in resisting this injury by reducing ROS generation (Ni et al., 2012). As we all know, mitochondria are capable of maintaining lipid homeostasis through fatty acid β-oxidation. Lin et al. (2013) reported that chloroquine, mitophagy inhibitor, induces liver damage, which features mitochondria injury-mediated liver steatosis, and rapamycin could attenuate this injury. It has also been found that mitophagy could reduce alcohol-induced liver injury (Ding et al., 2010a).
New perspectives on therapeutic strategies
As described previously, targeting dysfunctional mitochondria through activating mitophagy may be a novel perspective for therapy injured mitochondria-mediated diseases in the future. Although mitophagy may not thoroughly counteract the etiology of these diseases, it can potentially alleviate the symptomatology of them on account of its capability of improving mitochondrial function, resisting oxidative stress, and promoting cell survival.
In AD mouse models, long-term carbamazepine treatment decreases Aβ and amyloid plaque, and alleviates cognitive deficits since it has a role similar to rapamycin that enhances mitophagy by reducing mTOR activity (Caccamo et al., 2010; Li et al., 2013). In addition, rapamycin can reduce Huntingtin aggregation and cell death in HD cell models (Sarkar et al., 2009). Resveratrol, a promising drug for PD that induces mitophagy through AMPK/SIRT1 pathway, has neuroprotective effects against dopamine toxicity (Jin et al., 2008).
Ceramide can increase cancer cell death and chemotherapy sensitivity through inducing the formation of lipidated LC3 and then promoting mitophagy in cancer cells (Sentelle et al., 2012; Dany et al., 2016). Dihydroergotamine, another mitophagy inducer, was shown to inhibit the growth of lung tumor by decreasing membrane permeability, increasing ROS production as well as apoptosis, and disturbing ATP generation (Chang et al., 2016). Interestingly, mitophagy may also promote tumorigenesis during the late stage of tumor growth. It depends on cancer types and cellular circumstances and its mechanisms need to be further studied (Chourasia et al., 2015a; Kulikov et al., 2017).
The mitophagy enhancer spermidine exerts its antiaging protection on arteries by reducing oxidative stress and modifying structural factors (Eisenberg et al., 2016). Moreover, rapamycin was also found to significantly reverse cardiac aging proteome remodeling (Dai et al., 2014).
Conclusion
In this review, we summarize and discuss the cellular insults induced by excessive ROS, the mechanism of mitophagy, and the interactions between ROS and mitophagy in related stressful conditions, such as nutrient starvation, hypoxia, IR, and metabolic stress. ROS can cause cell injury through oxidative stress. However, mitophagy could conversely scavenge mitochondria impaired by ROS to maintain cellular homeostasis and promote cell survival. ROS are inevitable by-products of mitochondrial metabolism (Murphy, 2009), but, in response to stressful conditions, ROS can cause injury or even apoptosis for cells through the DDR (Shao et al., 2014), NF-κB (Hui et al., 2014), and P38 MAPK pathways (Xu et al., 2014). Several distinct mechanisms have been involved in mitophagy. For example, Atg32 is required to eliminate in yeast (Okamoto et al., 2009); Pink1/Parkin signaling (Xiao et al., 2017), NIX/BINP3 (Boyd et al., 1994; Ohi et al., 1999), and FUNDC1 pathways (Liu et al., 2012) are important for mitophagy in mammalian cells. Mitophagy could selectively eliminate ROS-impaired mitochondria to decrease ROS level and maintain its normal function. Moreover, ROS could also initiate mitophagy through various regulatory signaling pathways. For instance, ROS may reduce mitochondrial membrane potential, which initiates mitophagy through the Pink1/Parkin signaling pathway (Wang et al., 2012). In the nucleus, ROS could activate HIF-1, FOXO3, and NRF2, and they stimulate the transcription of BNIP3/NIX, LC3/BNIP3, and P62, respectively, consequently facilitating mitophagy (Jain et al., 2010; Shen et al., 2015; Klumpen et al., 2017). In brief, the interactions between ROS and mitophagy occur in various related pathological conditions. The internal regulatory mechanisms involved in ROS and mitophagy may provide a new angle on therapies for related diseases. However, such mechanisms need further investigation.
Footnotes
Disclosure Statement
No competing financial interests exist.