
Abstract
Salt response has long been considered a polygenic-controlled character in plants. Under salt stress conditions, plants respond by activating a great amount of proteins and enzymes. To develop a better understanding of the molecular mechanism and screen salt responsive genes in chrysanthemum under salt stress, we performed the RNA sequencing (RNA-seq) on both salt-processed chrysanthemum seedling roots and the control group, and gathered six cDNA databases eventually. Moreover, to overcome the Illumina HiSeq technology's limitation on sufficient length of reads and improve the quality and accuracy of the result, we combined Illumina HiSeq with single-molecule real-time sequencing (SMRT-seq) to decode the full-length transcripts. As a result, we successfully collected 550,823 unigenes, and from which we selected 48,396 differentially expressed genes (DEGs). Many of these DEGs were associated with the signal transduction, biofilm system, antioxidant system, and osmotic regulation system, such as mitogen-activated protein kinase (MAPK), Acyl-CoA thioesterase (ACOT), superoxide (SOD), catalase (CAT), peroxisomal membrane protein (PMP), and pyrroline-5-carboxylate reductase (P5CR). The quantitative real-time polymerase chain reaction (qRT-PCR) analysis of 15 unigenes was performed to test the data validity. The results were highly consistent with the RNA-seq results. In all, these findings could facilitate further detection of the responsive molecular mechanism under salt stress. They also provided more accurate candidate genes for genetic engineering on salt-tolerant chrysanthemums.
Introduction
T
Salt stress greatly activated the expression of AtMPK3 and AtMPK6 in Arabidopsis (Droillard et al., 2002). Overexpression of NHX1, a Na+/H+ antiporter, conferred salt tolerance in Arabidopsis and tobacco (Sottosanto et al., 2007; Zhou et al., 2008). Overexpression of several antioxidant enzymes such as GhSOD1 and GhCAT1 could increase the salt tolerance of plants (Luo et al., 2013). Besides, many transcription factors (TFs) had been identified the ability to improve the salt tolerance of plants, such as the overexpression of DREB1 enhanced the salt tolerance in transgenic Arabidopsis and rice (Oh et al., 2005; Zhang et al., 2009) and overexpression of WRKY11 conferred transgenic tobacco with higher salt and drought tolerance (Xu et al., 2014).
Chrysanthemum (Dendranthema grandiflorum) was one of the most famous cut flowers in the world, which had a huge annual demand. It was susceptible to salt stress (Wu et al., 2016). So far, research on the salt tolerance of chrysanthemum mainly focused on genetic engineering breeding. Many achievements had been made in the development of stress-tolerant transgenic chrysanthemums by transferring many kinds of TFs. For instance, overexpression of ClCBF1, ChiMYB, CmWRKY17, DgWRKY2, DgWRKY5, DgNAC1, AtDREB1A, and CgDREBa could improve the salt tolerance for chrysanthemum (Mie and Kazuko, 2006; Chen et al., 2011; Li et al., 2015; He et al., 2016, 2018; Wang et al., 2017; Liang et al., 2017; Gao et al., 2018). However, the salt tolerance mechanism in chrysanthemum was not further studied. How the signaling molecules genes, osmolytes, and ion transporters respond to salt stress in chrysanthemum was still unclear (Ahmad and Prasad, 2012). Therefore, the detection of salt response genes for tolerance breeding and the understanding of the salt response mechanism of chrysanthemum were of great importance.
The salt-responsive gene groups in chrysanthemum remained incomplete due to the enormous gene group of chrysanthemum. However, the development of sequencing technologies solved this conundrum (Thudi et al., 2012). Now, with the development of next-generation sequencing (NGS) technology, large-scale RNA sequencing (RNA-seq) had been conducted under various abiotic stresses in many species (Etika et al., 2016); Illumina HiSeq technology had become popular tool in studying known and discovering new genes for its high accuracy and sensitivity of gene discovery. Many salt-related differentially expressed genes (DEGs) had been identified by this technique in many species, such as rice (Oryza rufipogon Griff.), pear (Pyrus betulaefolia), wild barley (Hordeum spontaneum), cotton (Gossypium hirsutum L.), alfalfa (Medicago sativa L.), and radish (Raphanus sativus L.) (Yao et al., 2011a; Bahieldin et al., 2015; Sun et al., 2016; Zhou et al., 2016; Li et al., 2017; Lei et al., 2018). However, the full-length transcript obtained by Illumina HiSeq was incomplete for the insufficient length of read. The single-molecule real-time sequencing (SMRT-seq) technology from Pacific Biosciences (PacBio) platform had introduced powerful new tools to help solve this problem (Zhu et al., 2018). The sequencing length of the PacBio was 12–15k bp, which could cover various lengths of transcripts in eukaryotes. The obtained sequences were expected to be full-length or nearly full-length transcripts (Abdel-Ghany et al., 2016). The incomplete Illumina HiSeq's issue could be modified by SMRT-seq's complete reads, while SMRT-seq's inaccurate reads could be modified by more accurate data from Illumina HiSeq (Roberts et al., 2013; Xu et al., 2015). Compared with Illumina HiSeq's applications, the SMRT-seq had a narrower application range with limited reports (Dong et al., 2015; Hoang et al., 2017; Liu et al., 2017). The method of combining Illumina HiSeq and SMRT-seq has been applied to generate comprehensive information at the transcriptional level in recent years, which provides the scientific basis for perfecting the genome database and molecular breeding, such as barley (Hordeum vulgare L.), hot pepper (Capsicum annuum L.), switchgrass (Panicum virgatum L.), wild strawberry (Fragaria vesca), and salvia (Salvia miltiorrhiza) (Xu et al., 2016; Li et al., 2018; Ren et al., 2018; Zhu et al., 2018; Zuo et al., 2018). The study on salt stress-responsive mechanism and screening candidate genes by SMRT-seq technology had not been reported yet.
Since plant roots were the most vulnerable organs to soil salinity, we sampled the salt treatment group and the control group of chrysanthemum roots to identify the salt-responsive genes and further dissect the molecular mechanism underlying salt stress response by combining Illumina HiSeq and SMRT-seq analysis. Our study investigates gene expression changes induced by NaCl solutions (100 mM). Total RNA was extracted from roots of chrysanthemum after a 24-h salt stress treatment. DEGs in response to salt stress were identified and analyzed. Also, several salt stress-responsive genes were selected for quantitative real-time polymerase chain reaction (qRT-PCR) to verify the RNA-seq results. The obtained results could provide a deeper analysis of salt stress responsiveness in chrysanthemum, and the characterization of salt-induced genes could be used for breeding salinity tolerance into chrysanthemum.
Materials and Methods
Plant materials and salt treatment
The chrysanthemum var. jinba was used in this study. The buds raised from tissue-cultivated seedlings were grown on MS medium for 30 days and then were transferred to pots filled with a 1:1 mixture of peat and perlite for 3 days under the same condition in an illumination incubator (23°C ± 2°C, 16-h photoperiod). Plants were irrigated with 100 mM NaCl solutions until the water is saturated and overflows the pots, while the control group was irrigated with deionized water. The test solutions were applied every 6 h, with a volume of 300 mL. The roots were washed with deionized water after a 24-h treatment. About 0.5–0.8 g of young roots were collected from each plant as samples. Totally, 54 plants were involved as control and experimental groups, respectively. There were six samples (CK-1, CK-2, CK-3, T-1, T-2, and T-3) in total for RNA-seq and determination of physiological indexes. The experiment was performed thrice for accuracy.
Library preparation and sequencing
RNA was extracted by Trizol reagent (Invitrogen, Thermo Fisher Scientific, Inc.) for subsequent RNA-seq analysis. An amount of 1.5 μg RNA per sample was used for the RNA sample preparations. Six libraries were generated by NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB), following manufacturer's recommendations. The degree of RNA degradation and contamination was monitored on 1% agarose gels. The purity, concentration, and integrity of the libraries were detected by Nanodrop (IMPLEN), Qubit 2.0 (Life Technologies), and Agilent 2100 (Agilent Technologies), respectively. Then, the six libraries were sequenced by Illumina HiSeq 4000 platform.
The six samples of chrysanthemum roots were blended into a single sample. This sample was for PacBio library preparation. The library was sequenced by Iso-Seq with PacBio RS II systems (Pacific Biosciences). Raw data were submitted to NCBI sequence for reading and recording (digital log-in number is GSE114752).
Data processing and gene functional annotation
Raw data of Illumina HiSeq were processed through inner perl scripts. In this step, clean data were obtained by removing reads containing adapter, reads containing Ploy-N, and low-quality reads from raw data. The subreads were obtained from sequence data by SMRTlink 5.0. To identify full-length non-chimera (FLNC) sequence, we tested whether the circular consensus sequence (CCS) generated from subreads on the subject contained 5′-primer, 3′-primer, and Poly-A. With the data completed, the iterative clustering for error correction calculation was applied to cluster the FLNC sequences in the identical transcript. As a result, we obtained consensus sequences, which were modified by non-full-length sequences later. The polished consensus sequence was used in the following analysis. The CD-HIT software was used to remove redundancies and similar sequences from the combined product of consensus and unigenes by the 99% similarity standard (Fu et al., 2012). The procedure successfully delivered a nonredundant sequence file.
Gene function was annotated on the following databases: NCBI nonredundant protein sequences (Nr), NCBI nonredundant nucleotide sequences (Nt), protein family (Pfam), clusters of orthologous groups of proteins (KOG), a manually annotated and reviewed protein sequence database (Swiss-Prot), KEGG ortholog (KO) database, and gene ontology (GO).
Long noncoding RNA analysis
Coding–noncoding index (CNCI), coding potential calculator (CPC), Pfam and predictor of long noncoding RNAs (LncRNAs), and messenger RNAs based on k-mer scheme (PLEK) were used to predict the coding potential of genes (Kong et al., 2007; Fu et al., 2012; Sun et al., 2013; Li et al., 2014; Finn et al., 2016).
Differential expression analysis
Differential expression analysis between the control and experiment groups was determined by DESeq (Anders and Huber, 2010). The p value of the multiple-hypothesis test was modified by the False Discovery Rate. p Value <0.05 and |log2FC| >3 were recognized as significant differences in gene expression. GO enrichment and KEGG pathway enrichment analyses were performed by GOseq software and KOBAS (2.0), respectively (Mao et al., 2005; Young et al., 2010).
Validation of RNA-seq data by qRT-PCR
We selected 15 genes of different functions for qRT-PCR analysis to ensure the accuracy of the data of RNA-seq. The qRT-PCR was performed by the SsoFast EvaGreen supermix (Bio-Rad, Hercules, CA) and Bio-Rad CFX96™ detection system. A final 20 μL qRT-PCR reaction mixture contained 10 μL SsoFast EvaGreen supermix, 2 μL diluted cDNA sample, and 300 nM primers. Relative expression levels were calculated by the 2−ΔΔCT method, and the EF1α (Genbank Accession No. KF305681) was used as a reference for quantitative expression analysis. The whole experiment was conducted thrice for accuracy. The primers used in qRT-PCR are listed in Supplementary Table S1 (Supplementary Data are available online at
Determination of chrysanthemum's physiological indexes under salt stress
The contents of proline, soluble sugar (SS), glutathione (GSH), and hydrogen peroxide (H2O2), and the activities of superoxide anion (APX), peroxidase (POD), superoxide (SOD), and catalase (CAT) were measured by the Nanjing Jiancheng test kit. The content of superoxide anion (O2 −) was measured by the Suzhou Keming test kit. Both procedures were performed under the instructions. In addition, soluble protein (SP) content was determined by Irigoyen et al. (1992). A TGL-16G high-speed centrifuge (Shanghai Anting Scientific Instrument Factory) and an ultraviolet-visible (UV-Vis) spectrophotometer (Thermo Spectronic® 20 Genesys™) were applied to measure the physiological indexes.
Results
Overview of the RNA-seq
To obtain comprehensive transcriptome profiles of chrysanthemum roots under salt stress, six libraries were constructed under normal circumstance and salt stress. Totally, 45.56 Gb clean reads with an average length of 777 bp and N50 of 968 bp were produced by Illumina HiSeq 4000 (Supplementary Table S2).
For SMRT-seq, with a total of 6,472,969 subreads (10.84 Gb) scanned, 510,559 CCS reads and 365,261 FLNC reads appeared after data processing and screening. After the modification of site mismatch, a total of 204,507 consensus reads were acquired, with N50 of 2864 and average length of 2663 bp (Supplementary Table S3).
After the removal of redundancies, a total of 550,823 unigenes were collected with 52,120 ones over 3k bp of united sequencing, while the unigenes of Illumina Hiseq only had a focusing length range of 200–1k bp and 19,464 ones over 2k bp (Table 1).
SMRT-seq, single-molecule real-time sequencing.
Gene annotation and functional classification
To predict and analyze the function of the unigenes, we carried out functional annotation by applying a BLAST comparison with multiple databases. A total of 378,033 annotated genes were distributed to each of the databases, with 343,468 (90.85%) for Nr, 227,902 (60.29%) for KOG, 146,509 (38.76%) for GO, 326,571 (86.39%) for KEGG, 146,509 (38.76%) for Pfam, 284,965 (75.38%) for Swiss-Prot, and 182,633 (48.31%) for Nt (Supplementary Table S4).
The analysis on Nr database indicated the highest homology between chrysanthemum and Cynara cardunculus, with 160,655 (46.77%) unigenes annotated; followed by Anthurium amnicola and Daucus carota, with 4.54% and 2.74%, respectively (Fig. 1). A total of 146,509 unigenes were annotated successfully and classified into 57 functional groups of 3 GO main categories (biological process [BP], cellular component [CC], and molecular function [MF]). The top three GO terms for classified genes were “metabolic process” (69,698), “cellular process” (65,961), and “single-organism process” (40,838) for BP category; “cell” (27,987), “cell part” (27,987), and “organelle” (20,145) for CC; and “binding” (90,601), “catalytic activity” (68,490), and “transporter activity” (7779) for MF (Supplementary Fig. S1). A total of 326,571 unigenes were distributed into 379 pathways of KEGG database, the top three pathways were “Genetic Information Processing; Translation; Ribosome” (10,320), “Metabolism; Global and overview maps; Carbon metabolism” (6954) and “Metabolism; Global and overview maps; Biosynthesis of amino acids”(6358) (Supplementary Fig. S2).
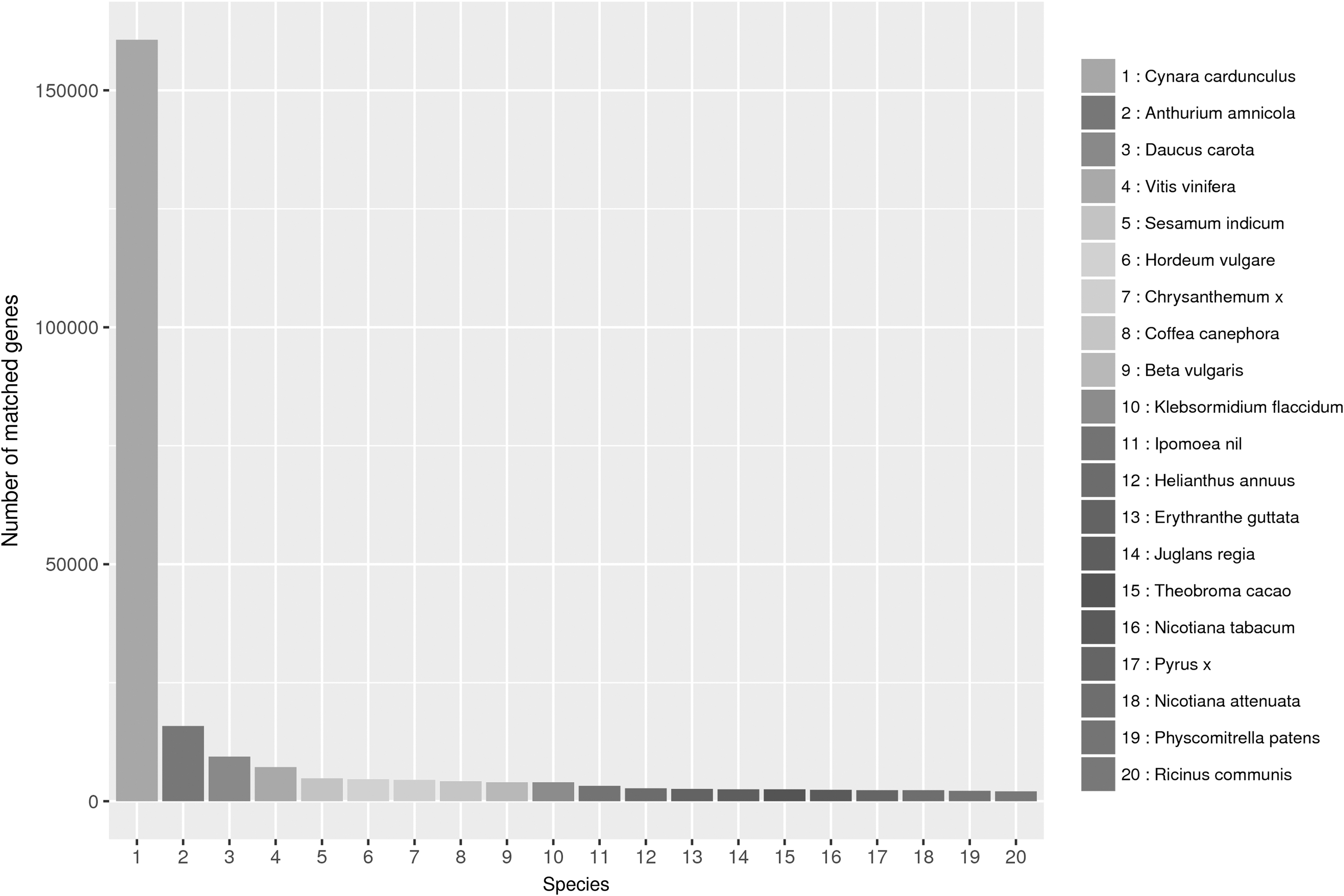
The species distribution of the NR annotation result. NR, nonredundant.
Forecasting for LncRNA
LncRNA is the RNA molecule of nonencoding protein and capable of adjusting the expression of genes at the epigenetic, transcriptional, and posttranscriptional level, participating in chromatin modification, sequence activation, transportations within nucleus, and so on. Many studies showed that LncRNA played an active role in both growth and threat response of a plant (Deng et al., 2018). All 4 software detected LncRNA with an amount of 463,613 (Fig. 2).
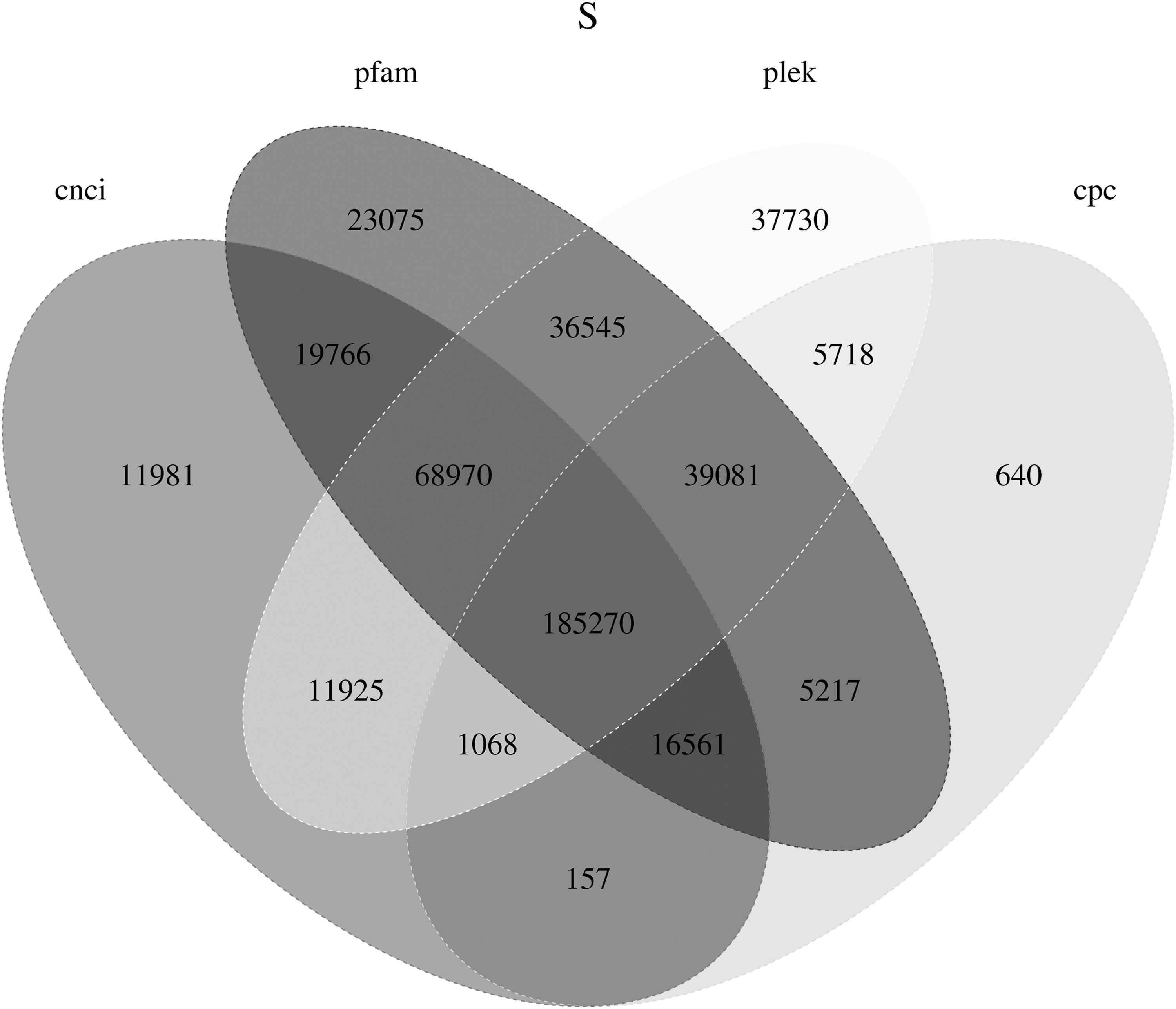
Venn diagram of the predicted LncRNA. LncRNA, long noncoding RNA.
Analysis of DEGs
To investigate the gene expression patterns of chrysanthemum's root under salt stress, fragments per kilobase of exon per million fragments mapped (FPKM) values were used to normalize the reads from RNA-seq. Thus, the gene expression was compared between the control and experimental groups. Among all 48,396 DEGs, 38,822 (80.22%) of them were upregulated, while the other 9574 (19.78%) were downregulated. Finally, a total of 35,874 DEGs were annotated by 7 databases: NR (30,316), KOG (24,585), GO (14,739), KEGG (27,917), Pfam (14,739), Swiss-Prot (29,505), and Nt (11,005) (Supplementary Table S4).
As an effort to discover the relativeness between DEGs and its products, we performed the GO enrichment analysis; 14,739 annotated DEGs were included into the GO database through the process. The top three GO terms for classified genes were “nitrogen compound metabolic process,” “cellular nitrogen compound metabolic process,” and “biosynthetic process” for BP; “cell,“cell part,” and “intracellular” for CC and “structural molecule activity,” “structural constituent of ribosome,” and “RNA polymerase II transcription factor activity, sequence-specific DNA binding” for MF (Supplementary Fig. S3).
In addition, there were 10 essential nodes discovered in the BP, CC, and MF. The most noticeable one in BP was “nitrogen compound metabolic process” and in CC was “ribonucleoprotein complex,” while two significant nodes in MF were “structural molecule activity” and “structural constituent of ribosome” (Supplementary Table S5).
A total of 27,917 DEGs were assigned to the KEGG database and involved 125 pathways; the top 20 significantly enriched pathways of DEGs are presented in Supplementary Table S6, which were closely related to plant growth and development.
DEGs responding to salt stress in chrysanthemum
The response of DEGs to salt stress is a primary process for plants to activate defense system. There were two acyl-CoA thioesterase (ACOTs) related to the biofilm system significantly upregulated. A total of 76 DEGs related to ROS scavenging were identified under salt stress, including SOD, CAT, peroxisomal membrane protein (PMP), and glutathione s-transferase (GST) genes. Totally, 3 pyrroline-5-carboxylate reductase (P5CR), eleven 6-phosphogluconate dehydrogenase (6PGDH), 7 fructose 1,6-bisphosphatase (FBP), 19 malate dehydrogenase (MDH), and 9 hexokinase (HK) genes related to osmotic regulation system were differentially expressed under salt stress. Moreover, in our study, 23 DEGs related to ion transporters were identified, including Na+/H+ antiporters and nitrate transporters (NRTs).
A total of 51 DEGs of calcium-dependent protein kinase (CDPK) family, 28 DEGs of calcineurin B-like interacting kinase (CIPK) family, and 5 calcineurin B-like (CBL) proteins were found associated with signal transduction. Besides, totally, 22, 12, and 28 genes involved in the mitogen-activated protein kinase (MAPK), MAP2K, and MAP3K cascades were also identified related to signal transduction, respectively.
In addition, our analysis proved that other types of DEGs might be related to salt stress response in chrysanthemum, such as heat shock proteins (HSPs), late embryogenesis abundant (LEA) proteins, branched-chain amino acid aminotransferase (BCAT), thiamine pyrophosphokinase (TPK), and thiamine thiazole synthase (THI4) genes.
All 119 DEGs belonging to HSP family enriched in “Spliceosome” pathway under salt stress, most of them showed a significant upregulation. We speculated that HSPs participated in the progress of alternative splicing for the chrysanthemum's root and structure stabilization for protein and cytomembrane under salt stress, and effectively reduced the damage caused by salt stress. Similarly, our data discovered that 13 LEA proteins were differentially expressed and that the majority of them were upregulated. This indicated that these genes might play a vital role in the reaction of chrysanthemum's root cell under salt stress.
The majority of these DEGs were upregulated and the discovery of these genes might provide new research resources for the breeding of salt-tolerant chrysanthemums. All DEGs responding to salt stress are shown in Supplementary Tables S7–S16 and part of them were listed in Table 2.
6PGDH, 6-phosphogluconate dehydrogenase; ACOT, acyl-CoA thioesterase; BCAT, branched-chain amino acid aminotransferase; CAT, catalase; FBP, fructose 1, 6-bisphosphatase; GST, glutathione s-transferase; HK, hexokinase; HSPs, heat shock proteins; MDH, malate dehydrogenase; NRTs, nitrate transporters; P5CR, pyrroline-5-carboxylate reductase; PMPs, peroxisomal membrane proteins; SOD, superoxide; THI4, thiamine thiazole-synthase; TPK, thiamine pyrophosphokinase.
TFs responding to salt stress
Once suffering environmental stresses, TFs could function as significant molecular switches on specific responsive genes. In this study, totally 1585 (1043 upregulated and 542 downregulated) TFs were identified. Among them, the majority of TFs belonging to zn-clus (143), C2H2 (130), bZIP (65), SNF2 (61), GNAT (47), C3H (42), and bHLH (40) families were predominantly upregulated, indicating their critical involvement in salt stress response (Fig. 3).
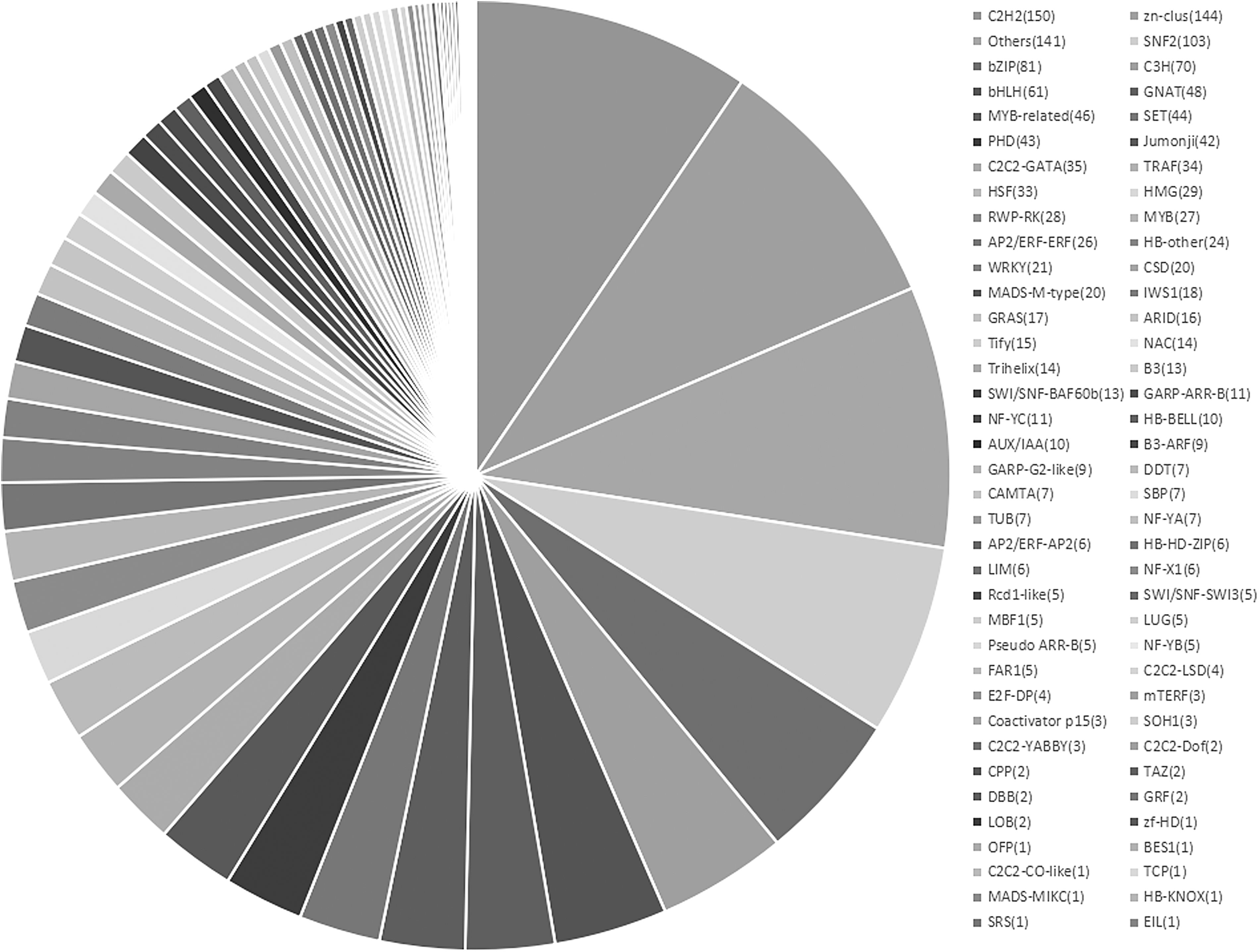
Differentially expressed TFs under salt stress. TFs, transcription factors.
DEGs' verifications by qRT-PCR
To verify the gene expression level gathered from united sequencing, we randomly selected 15 DEGs for qRT-PCR verification, including MYB, GRAS, bZIP, WRKY TF, MAPK, Cu/ZnSOD, RNA replication protein, NADH dehydrogenase, Protein phosphatase 2C, HSP, Na+/H+ antiporter, FBP, GST, GTP-binding protein, and LEA protein. The result stated a strong similarity with RNA-seq data (Fig. 4).
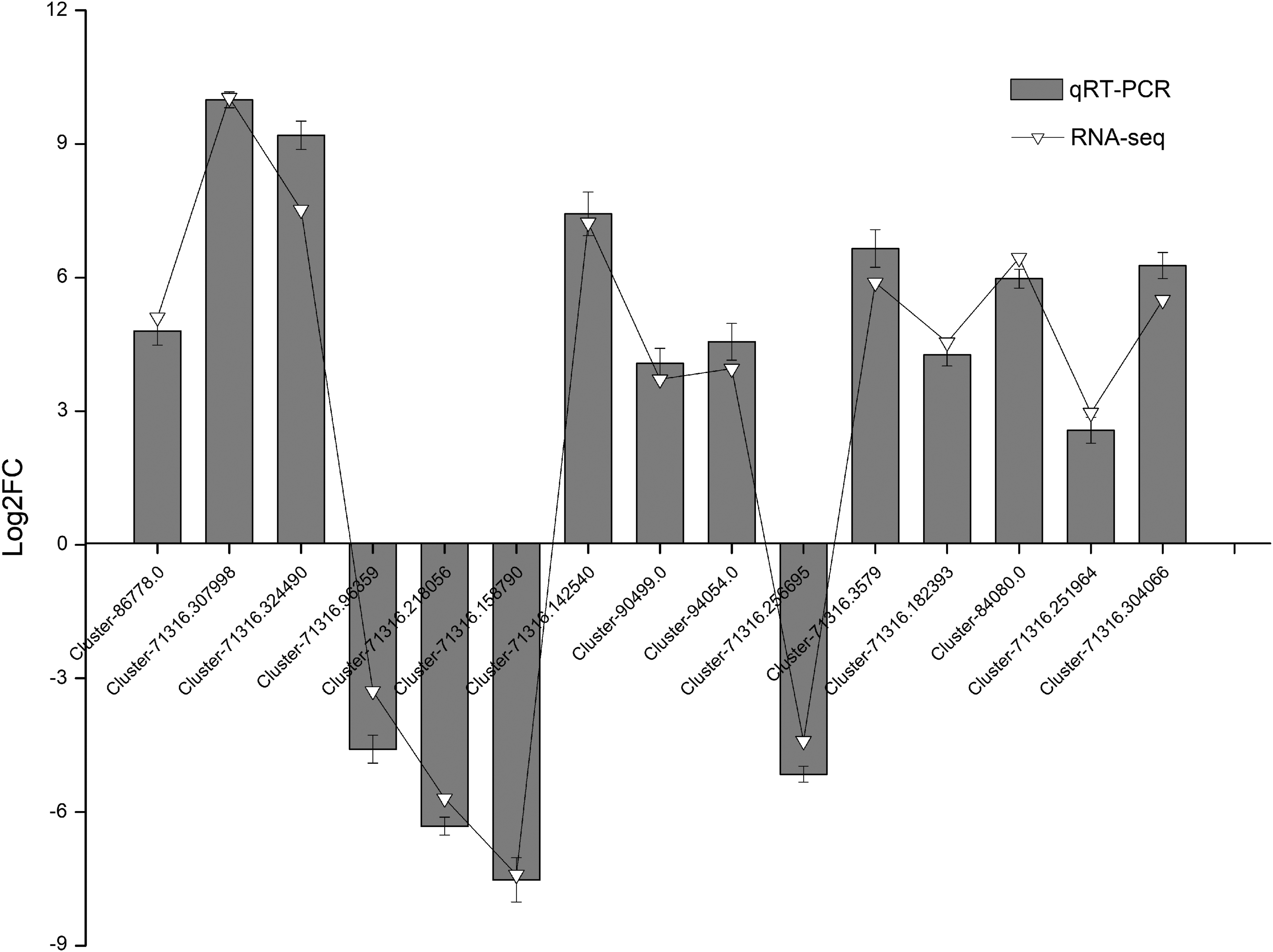
The relative expression levels of 15 DEGs between RNA-seq and quantitative real-time polymerase chain reaction (qRT-PCR). The gene relative expression levels were determined by 2−ΔΔCT as expressed, and were normalized to the expression level of EF1α. All samples were run in triplicate. RNA-seq, RNA sequencing.
Analyses of phenotypic and physiological changes of chrysanthemum under salt stress
As it showed in Figure 5A, the phenotype of chrysanthemum showed no manifest difference from normal circumstances. However, the treatment group accumulated more H2O2 and O2 − than the control group in chrysanthemum roots (Fig. 5B, C). The contents of MDA, GSH and the activities of antioxidizes (APX, POD, SOD, and CAT) in the treatment group were remarkably higher than those that under normal conditions. Besides, an observable increase appeared in osmotic adjusting materials under salt stress, such as proline, SS, and SP (Fig. 6).

Phenotypic and physiological changes of chrysanthemum under salt stress. CK is the control group and T is the treatment group.

Physiological indexes of chrysanthemum under salt stress. CK is the control group and T is the treatment group.
Comparison analysis between Illumina HiSeq data and SMRT-seq data
A lot of unigenes from Illumina HiSeq data were successfully mapped to genes from SMRT-seq data. For example, the gene ID Cluster-1259.1 was mapped to SMRT-seq data and modified for 291 sites, while the length was increased to 1181 bp. Cluster-71316.291135 was modified for 111 sites and increased by 1444 bp.
In the comparison analysis, we discovered that most genes' position points were modified by the united sequencing. At the same time, the length of genes increased. This improvement allowed us to obtain a more accurate open reading frame, which enhanced the accuracy of the transcriptomes' data (before-and-after comparison between 18 genes is listed in Table 3).
Discussion
Salt, as an important stress factor, severely affected the growth of plants as well as casting a severe shadow on the metabolic reaction (Wu et al., 2016). “Jinba” exactly is a kind of cutting-chrysanthemum, which is difficult to mass produce under salt damage.
With the development of RNA-seq, the available data of chrysanthemum's salt response are no longer limited. Combining Illumina Hiseq with SMRT-seq, more and more precise candidate genes were detected. SMRT-seq genes were longer than Illumina Hiseq unigenes, and many Illumina Hiseq unigenes could be mapped to SMRT-Seq genes, with the modification and enlargement of the length. Since most candidate genes assembled from the Illumina Hiseq reads did not well-represent full-length transcript, our united sequencing data improved the quality of transcripts assembled from Illumina short reads and significantly improved the appraising efficiency of full-length transcripts.
In the analysis of DEGs, we unveiled mass of salt-responsive genes and the majority of them were involved in upregulation rather than downregulation. For instance, BACT could promote the degradation of branched amino acids (BCAAs), such as leucine, isoleucine, and valine, the essential amino acid for sugar production. The degradation of BCAAs enhanced cells' movements since they supplied cells with sugar and energy (Letto et al., 1986; Hutson et al., 1995). We found that 10 BCAT genes expressed a significant upregulation. The BCATs could be closely related to chrysanthemum's response to salt stress.
It was worth noticing that, Rapala-Kozik et al. (2008) discovered that thiamine metabolism played an essential role when corn seeds reacted to abiotic stress. Particularly, the TPK increased under drought, high salt, and oxidative stresses. Orthologues of the yeast THI4 gene that controlled a single step were upregulated under conditions of abiotic stresses (Machado et al., 1997). In our research, 1 TPK and 11 THI4 genes were identified and significantly upregulated, which could be essential for the study of thiamine metabolism of chrysanthemum under salt stress.
Salt-responsive genes associated with the biofilm system
Biomembranes were not only the primary natural barrier between a creature itself and the outside environment but also the main damaged site in the reaction to environmental stress (Foyer and Shigeoka, 2011). Under salt stress, membrane lipid peroxidation caused cell membrane damage and destroyed membrane lipid mobility. As the main product of membrane lipid peroxidation, MDA was widely used in measuring the degree of membrane lipid overoxidation (Yoshimura et al., 2004). The MDA content in chrysanthemum's root stated a significant upward tendency when salt condition occurred (Fig. 6A). Unsaturated fatty acids could increase membrane fluidity. The ACOT gene promoted the synthesis of α-linolenic acid and played a crucial role in response to salt stress by adjusting the degree of unsaturation of fatty acids in Chlamydomonas nivalis (Jiang and Wei, 2010). Tan et al. (2014) found that CoACOT, an auxiliary gene, could regulate and maintain the physiological balance in lipid synthesis of Camellia oleifera seeds. Hunt et al. (2002) found that an ACOT could function as a key regulator of peroxisomal lipid metabolism. The significant upregulation of the ACOT gene indicated that chrysanthemum responds to salt stress by synthetizing unsaturated aliphatic acid.
Salt-responsive genes associated with antioxidant system
Salt stress induces ROS, which leads to secondary oxidation stress, disturbs cellular redox homeostasis, and damages cell components and structures. The alleviation of oxidative damage and increased tolerance to salt stress are often correlated with the balance between ROS producing and ROS scavenging (Baxter et al., 2014). Peroxisome could remove H2O2 and O2 − from the plants and functioned in the dynamic adjustments to ROS (Mittova et al., 2003). The increase of SOD and CAT spared plants from the harm of reactive oxygen, for they were the plant's cleaning agents (Mahmut et al., 2005). It has been reported that SOD1 and CAT1 acted as ROS scavengers and participated in salt stress in cotton (Luo et al., 2013). The PEX11 family of peroxisome membrane proteins had been involved in the regulation of size and number of peroxisome in plant, animals, and yeast cells. Recently it was reported that several rice PEX11 genes were upregulated under abiotic stress (Mitsuya et al., 2010). Cui et al. (2016) found that OsPEX11 contributed to salt stress tolerance in Oryza sativa. GSTs could reduce the amount of reactive oxygen produced in stressed environment. The overexpression of GST genes proved to promote salt tolerance in Arabidopsis, rice, and tobacco (Roxas et al., 1997; Jain et al., 2010; Qi et al., 2010). Ji et al. (2010) introduced the wild soybean GST gene into tobacco, which significantly improved the drought and salt tolerance of transgenic tobacco.
Under salt stress, more H2O2 and O2 − were accumulated in the roots of chrysanthemum (Fig. 5B, C). The ROS scavengers showed a significant upward trend (Fig. 6B–F); the expression of these responsive genes notably upregulated when salt condition occurred. These results suggested that the plant defense system eliminated secondary ROS damages elicited by salt stress.
Salt-responsive genes associated with osmotic regulation system
Osmoregulation played a crucial role in mitigating salt-triggered damage under osmotic stress (Liu et al., 2018). It is well proposed that the accumulation of compatible osmolytes in the cytoplasm, such as proline and sugars, plays a pivotal role in lowering the osmotic potential and thus promotes salinity tolerance of plants (Adams et al., 1998). P5CR could transfer pyrroline-5-carboxylate back to proline. It has been reported that overexpression of TaP5CR in transgenic Arabidopsis improved its tolerance to salt stress. Overexpression of IbP5CR enhanced salt tolerance in transgenic sweet potato. The increase of proline content is also found in the TaP5CR-overexpressing Arabidopsis and the IbP5CR-overexpressing sweet potato (Ma et al., 2008; Liu et al., 2014). Thus, we believed that P5CR genes' inducing expressions increased the proline content in the chrysanthemum under salt stress. This functioned in the reaction to a stable osmotic pressure, preventing further harm from osmotic stress.
Sugar was an essential regulator in cell osmosis. The synthesis and decomposition of sugars affected the osmotic potential of cells (Rolland et al., 2005). We discovered many upregulated enzymes in carbohydrate metabolism pathways, such as 6PGDH, FBP, MDH, and HK. It has been proved that 6PGDH, FBP, MDH, and HK participated in the adjustment progress against nonbiological stress. In fact, 6PGDH was one of the key enzymes in the pentose phosphate pathway. FBP was a key enzyme in the synthesis of sucrose. MDH was a key enzyme in the citrate cycle and HK was the key enzyme in the glycolysis pathway (Strand et al., 2000; Yao et al., 2011b; Yang et al., 2014; Jin et al., 2016). Os6PGDH was upregulated in the shoots under salt stress and played an important role in cell division and salt response (Huang et al., 2003). FBP activity in the leaves of salt-stressed rice plants increased for sugar biosynthesis and accumulation (Chaum et al., 2009). Overexpression of MdcyMDH gene enhanced the ability of cold and salt tolerance on apple's callus and tomato (Yao et al., 2011b). HK could regulate glycolytic activity in maize root tips and tomato roots (Bouny and Saglio, 1996; Germain et al., 1997). The significant upregulation of these genes illustrated that salt-stressed chrysanthemum could defend itself by adjusting the activeness of these key enzymes against osmotic stress.
Organic osmolytes are effective osmoprotectants alleviating osmotic damage imposed by salt stress (Borochov et al., 1986; Yasseen et al., 2006). As the most common osmotic pressure regulators in salt-stressed plants, proline, SS, and SP contents exhibited a significant increase (Fig. 6G–I). The notable upregulation of P5CR and enzymes related to carbohydrate metabolism facilitated the accumulation of compatible osmolytes and mitigated salt-elicited osmotic damage.
In addition, the moderate absorption of some active inorganic ions significantly contributed to osmotic adjustment under salt stress. It accompanied by ion toxicity, most of the time (Chen and Jiang, 2010). This occurred with the strong expression of various types of ion transporters, such as Na+/H+ antiporters and NRTs.
Once Na+ enters the cytosol, it can potentially be excluded (back to the soil) by Na+/H+ exchangers located in the plasma membrane, or sequestered into the vacuole by Na+/H+ exchangers located in the tonoplast to maintain osmotic balance (Apse et al., 1999). Recently, the primary functions of Na+/H+ antiporters in response to salt stress have been demonstrated by overexpressing Na+/H+ antiporter genes in transgenic Arabidopsis, tomato, and rice (Zhang and Blumwald, 2001; Ohta et al., 2002; Shi et al., 2002). The significant expression of Na+/H+ antiporter is beneficial to reconstitute the cytosolic K+/Na+ homeostasis and to improve the plant's salt tolerance (Sun et al., 2016).
For most plants, nitrate was the major source of nitrogen. That nitrate entered root cells was the first step of the NO3 - assimilation pathway. This was considered a key step in nitrogen metabolism (Kant, 2017). Popova et al. (2003) found that McNRT1 in Mesembryanthemum crystallinum had an upregulated expression when reacting to salt stress. Chopin et al. (2007) demonstrated that ATNRT2.7 played a specific role in nitrate accumulation in the seed. Such results might support that chrysanthemum could transport nitrate to enhance its self-protecting ability against salt stress.
Taken together, ion transporters play key roles in the regulation of salt stress response in chrysanthemum; it could be speculated that osmoregulation was a complex process and contributed to mitigate salt damage in chrysanthemum.
Signal transportation under salt stress
Plants are able to sense and transduce the stress signals to activate a set of defense mechanisms when exposed to salt stress. This process requires the involvements of numerous salt-related genes that serve as activators and regulators of stress response (Ahmad and Prasad, 2012). The results showed that numerous genes implicated in calcium signaling pathway and MAPK signaling pathway were significantly differentially expressed in salt-treated chrysanthemum roots compared to the control.
In plants, calcium functions as a ubiquitous secondary messenger in response to various environmental stresses (Bergey et al., 2014). Aside from this signaling molecule, the calcium signaling system also possesses other key components, including Ca2+ signaling sensors (e.g., CBLs and CDPKs) and decoders (e.g., CIPKs). Overexpression of AtCIPK6 gene in Arabidopsis increased plant tolerance to salt stress, AtCPK23 played an important role in Arabidopsis responses to drought and salt stresses, and CBL10 played a unique role in the Arabidopsis salt stress response (Ma and Wu, 2007; Quan et al., 2007; Chen et al., 2013). In this study, 51 CDPK, 28 CIPK, and 5 CBL genes were differentially expressed under salt stress, suggesting their specific roles in transducing salt stress signal.
Noteworthy, it was well proposed that under salt stress in Arabidopsis, increasing free cytoplasmic calcium could initiate the SOS pathway that is mainly attributed to the activation of Na+/H+ antiporter, and thereby reinstated cellular ion homeostasis (Qiu et al., 2003). In our study, 17 Na+/H+ antiporters were found, indicating the consociation of two pathways in salt stress response in chrysanthemum.
Previous studies have reported that some MAPK cascade genes can regulate salt stress tolerance in many plants (Yong and Min, 2010). Salt stress induced the expression of MAPK genes, such as AtMPK4 in Arabidopsis, and ZmMPK3, ZmMAPK5, and ZmSIMK1 in corn (Droillard et al., 2004; Ding et al., 2009). Overexpression of OsMAPK33 and OsMKK6 enhanced sensitivity to salt stress in rice (Lee et al., 2011). Overexpression of MAP3K conferred salt tolerance in Arabidopsis (Matsuoka et al., 2013). In addition, some studies also declared that MAPKs could modulate hormone signal transduction pathway to respond to stress damages. For instance, MPK6 was proven to regulate the productions of ethylene and jasmonic acid under stress (Liu and Zhang, 2004; Takahashi et al., 2007). As expected, 22 MAPK, 12 MAP2K, and 28 MAP3K genes were induced by salt stress, suggesting that the MAPK signaling pathway is of significance in salt responsive in chrysanthemum.
TFs implicated in salt stress response
TFs are well known to play essential roles in response to various abiotic stresses by regulating specific downstream genes (Wang et al., 2016). Salt stress can induce and depress the expressions of many TFs. Shen et al. (2014) identified 33 TFs as salt-inducible genes in Arabidopsis, including members of the AP2/ERF, MYB, bHLH, and NAC families. In our research, a large number of TFs took part in chrysanthemum's adjusting mechanism against salt stress. Among the common TF families, C2H2 and bZIP had greater amount of upregulated factors and followed by zn-clus, SNF2, and GANT, which were originated from uncommon families.
Among all TFs from these families, BnbZIP2, ThbZIP1, and AtbZIP17 act as positive regulators in plants' responses to high salinity stress (Liu et al., 2010; Wang et al., 2010; Huang et al., 2016). StZFP1, a C2H2-type zinc finger protein gene, involved in potato responses to salt and dehydration stresses through an ABA-dependent pathway (Tian et al., 2010). Overexpression of ZFP182 increased salt tolerance in transgenic rice (Huang et al., 2007). SNF2 has proven resisting ability toward abiotic stresses in model plants, such as rice and Arabidopsis (Li et al., 2011; Han et al., 2012), while the ability of other TFs such as zn-clus and GANT to relate to salt stress remains to be studied.
The regulation of TFs belonging to different families reflected the complexity of gene regulatory network linking to salt stress response in chrysanthemum (Hoang et al., 2014). Generally, it could be inferred that TFs played central roles in salt-responsive regulatory network in chrysanthemum.
Conclusion
In this study, we presented a united RNA-seq analysis focusing on chrysanthemum roots under salt stress. A total of 550,823 unigenes and 48,396 DEGs were identified. Based on the results, we explored series of potential salt-responsive genes related to signal transduction, biofilm system, antioxidant system, and osmotic regulation system. The majority of these genes, which also functioned in other plants under salt stress, were upregulated. The results provided a deeper insight into the molecular mechanism of chrysanthemum in response to salt stress, and successfully collected diversified data of chrysanthemum's full-length transcripts. The identified candidate genes facilitated the future studies on genetic engineering and breeding of chrysanthemum.
Footnotes
Acknowledgment
This research was supported by National Natural Science Foundation of China (31770742).
Authors' Contributions
Q.Z. and Q.L.L. conceived and designed the experiments. Q.Z., L.H., and B.W. performed the experiments. Q.Z., Y.Z.P., B.B.J., L.Z., F.Z., G.L.L., and Y.J. analyzed and interpreted the sequence data. Q.Z. wrote the article. All authors read and approved the article.
Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.