
Abstract
This study was aimed to identify hub genes associated with the development of glioblastoma (GBM) by conducting a bioinformatic analysis. The raw gene expression data were downloaded from the Gene Expression Omnibus database and The Cancer Genome Atlas project. After the differentially expressed genes (DEGs) were identified, the functional enrichment analysis of DEGs was conducted. Subsequently, the protein–protein interaction (PPI) network, molecular complex detection clusters, and transcriptional factor (TF)-miRNA-target regulatory network were constructed, respectively. Furthermore, the survival analysis of prognostic outcomes and genes was analyzed. In addition, the expression of key genes was validated by quantitative real-time PCR (qRT-PCR) analysis. A total of 884 DEGs, including 418 upregulated and downregulated genes, were identified between GBM and normal samples. The PPI network comprised a set of 3418 pairs involving 751 nodes, and AKT1 and CDK2 were the critical genes in the network. A total of seven clusters were identified, the genes in which were intensively associated with cell cycle, cholinergic synapse, and extracellular matrix (ECM)-receptor interaction. qRT-PCR analysis indicated that AKT1 and CDK2 were significantly upregulated, and NRXN3 and NPTX2 were significantly downregulated in GBM samples. The TF-miRNA-target regulatory networks were built, in which CCNB1, RFC5, microRNA524, and microRNA34b were key regulators. There were 43 genes, including NPTX2 and NRXN3, significantly related to the prognostic outcomes of GBM patients. These crucial genes might be promising options for GBM treatment.
Introduction
Gliomas, known as the most common primary malignancies in the brain, are classified as either relatively slow growing low-grade (I or II) tumors or rapidly growing, highly metastatic high-grade (III or IV) tumors (Charles et al., 2012). Glioblastoma (GBM) belongs to grade IV glioma and it is a catastrophic disease for both patients and their family. At present, the standard clinical treatment is surgical resection of the malignant tissues combined with radiotherapy and chemotherapy (Minniti et al., 2013; Oike et al., 2013). Despite the available therapies, the prognosis of GBM patients is extremely poor. Generally, the majority of patients do not survive for >2 years with a median survival of about 15 months and the 5-year overall survival (OS) rate remains <10% (Alcedo-Guardia et al., 2016; Nørøxe et al., 2017). The high resistance to therapeutics and the high variability of the effect of therapeutic drugs in different GBM patients take key roles for poor prognosis (Carlsson et al., 2010). Single-agent targeted therapy is usually inefficacious for GBM, while targeted drugs combined with epigenetic or immunomodulatory approaches may be used to combat the tumor (Chen et al., 2016; Touat et al., 2017). Therefore, it is necessary to reveal the pathogenesis of GBM to obtain the candidate therapeutic targets.
Genomic profiling analyses of GBM have been focused to identify potential new treatment options targeting related molecules and signaling pathways. Multiple candidate genes, such as soluble tumor necrosis factor receptor-1 (Ahluwalia et al., 2016), vascular endothelial growth factor (VEGF) (Reardon et al., 2008), and fibroblast growth factor (FGF)-2 (Ohashi et al., 2014), have been shown to be associated with the prognosis of GBM, and are considered potential therapeutic targets. Indeed, molecules targeting VEGF have been applied in the treatment of GBM. Bevacizumab is a humanized monoclonal antibody against VEGF and it was conditionally approved to treat the recurrent GBM in 2009 in the United States (Cohen et al., 2009; Lu-Emerson et al., 2015). A2V, an Ang-2/VEGF bispecific antibody against both Ang-2 and VEGF, can reprogram the tumor immune microenvironment, delay the growth of tumor, and prolong survival in GBM mouse model (Kloepper et al., 2016). The abnormal gene expression profiles are driven by microRNAs (miRNAs or miRs), transcriptional factors (TFs), and other regulators. For instance, miR-186-5p, a tumor suppressor miR, inhibits the tumorigenesis of GBM by directly targeting mRNAs of both FGF-2 and NF-κB subunit RelA (Wang et al., 2017). The driver factors and their affecting dysregulated genes induced dysregulated networks that control the development of GBM.
Microarrays, which are valuable tools for identifying disease-related genes, have been used to screen key molecules in GBM (Oike et al., 2013; Chen et al., 2016; Touat et al., 2017). In the present study, the GSE31262, a GBM-related gene expression profile data set provided by Sandberg et al. (2013), in which the gene expression was compared between GSCs (glioma stem cells) and ahNSCs (adult human neural stem cells) harvested from the subventricular zone, from the Gene Expression Omnibus (GEO) database and mRNA-seq data from The Cancer Genome Atlas (TCGA) project, was downloaded and analyzed to identify the dysregulated function and TF-miRNA-target networks in GBM. Besides, we extracted the clinical outcomes of GBM patients from the TCGA project and conducted survival analysis to identify critical genes related to prognosis. Finally, the expression of key genes was checked by quantitative real-time PCR (qRT-PCR) experiments. We aimed to show partial molecular mechanisms responsible for GBM, and to find promising molecule agents as therapeutic candidates of GBM patients.
Materials and Methods
Data acquisition
The raw gene expression data related to GBM, which were uploaded by Sandberg et al. (2013), were downloaded from the GEO database 1 with the accession number GSE31262. A total of 14 biopsy specimens collected from the informed and consenting patients were analyzed, including five individual samples of adult neural stem cells and nine individual samples of GSCs, and the gene expression profile data were collected based on the GPL2986 ABI Human Genome Survey Microarray Version 2. The tissue harvesting in their study was approved by the Norwegian National Committee for Medical Research Ethics.
The mRNA-seq data and the clinical data were obtained from TCGA project which were shown in FireBrowse. 2 Data of 152 GBM or para-carcinoma samples and 5 normal samples were extracted and analyzed.
Differential expression analysis
The limma (Smyth et al., 2005) package in R was used to analyze the gene expression profile of GBM from the GEO database and identify the genes with abnormal expression. The p-value was adjusted by multiple tests using the Benjamini–Hochberg (BH) procedure (Benjamini and Hochberg, 1995). The genes with adjusted p < 0.05 and |log fold change (FC)| > 0.58 were identified as the differentially expressed genes (DEGs) between GBM and normal patients.
The mRNA Seq data were preprocessed using the edgeR package (Robinson et al., 2010) (Version: 3.4). 3 First, a linear model was built among the normalized data (TMM normalization) and the average variance was adjusted by the precision weight calculated by the voom function. Then, the differential expression between tumor samples and normal samples was analyzed using the t-test in limma package. Finally, the p-value was adjusted in the same way as above. The adjusted p < 0.05 and |log FC| > 0.58 were set as the threshold values.
The overlapping DEGs were selected as the hub genes of GBM.
Functional enrichment analysis
The overrepresented Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway among the up- and downregulated genes were analyzed using the DAVID (Huang et al., 2009) (version: 6.7) tool. 4 Those enriched with more than 2 DEGs and with the hypergeometric p-value <0.05 were selected as the significant ones.
Construction of protein–protein interaction network
The protein–protein interactions (PPIs) among the DEGs were analyzed based on the STRING database (Szklarczyk et al., 2015) (Version: 10.0), 5 with the default parameters, and a combined score of >0.4 was set as the threshold. Then, the PPI network was constructed and Cytoscape software (Shannon et al., 2003) was used to visualize this network. Finally, topological properties of the PPI network were analyzed by the CytoNCA plug-in (Tang et al., 2014) (Version 2.1.6), 6 without weight. The data, including degree centrality, betweenness centrality, and closeness centrality, were obtained, based on which the scores of every DEG in the network were calculated and the top 10 genes with the highest scores were considered the principal ones. Besides, the Molecular Complex Detection (MCODE) algorithm (Bader and Hogue, 2003) was applied to detect the densely connected regions among the above large PPI networks. The MCODE clusters with score >5 were extracted and subjected to KEGG pathway enrichment analysis. The BH-adjusted p-value <0.05 was set as the cutoff value.
Construction of the TF-miRNA-target regulatory networks
To further explore the regulation of DEGs, we screened the possible miRNAs and TFs. Predication of the miRNAs targeting genes in the PPI network was conducted using the WebGestalt tool 7 (Zhang et al., 2005; Wang et al., 2013). A gene count ≥5 and the hypergeometric and BH-adjusted p-value <0.05 were considered significant. The TFs, which also are responsible for the regulation of DEGs, were collected from the ITFP website and the TRANSFAC professional database. 8
Thereafter, the networks, comprising these genes, corresponding TFs, and targeted miRNAs, were constructed.
Relationship between the differential mRNAs and prognostic outcomes
The median expression values of every differential mRNA in tumor samples were calculated, based on which they were grouped into high (above median)- and low-expression (below median) sets. Integrated with the prognostic outcomes, including the OS and the vital status, obtained from TCGA database, the Kaplan–Meier survival curves of these two groups were plotted and the log-rank tests were used to assess the relationship of gene expression to survival. A p-value <0.05 was set as the threshold for statistical significance.
qRT-PCR analysis
For qRT-PCR experiments, five individual samples of adult neural stem cells (from donated tissues) and five individual samples of GSCs (from the excisional tissues during surgery) were isolated according to previous literature (Vik-Mo et al., 2010). This study got the approval of the Ethics Committee of China-Japan Union Hospital of Jilin University, and received informed consent from participants.
The primer sequences for AKT1 (serine/threonine kinase 1; forward primer: 5′-TCCTCCTCAAGAATGATGGCA-3′, and reverse primer: 5′-GTGCGTTCGATGACAGTGGT-3′), CDK2 (cyclin-dependent kinase 2; forward primer: 5′-GTACCTCCCCTGGATGAAGAT-3′, and reverse primer: 5′-CGAAATCCGCTTGTTAGGGTC-3′), NRXN3 (neurexin 3; forward primer: 5′-AGTGGTGGGCTTATCCTCTAC-3′, and reverse primer: 5′-CCCTGTTCTATGTGAAGCTGGA-3′), and NPTX2 (neuronal pentraxin II; forward primer: 5′-ACGGGCAAGGACACTATGG-3′, and reverse primer: 5′-ATTGGACACGTTTGCTCTGAG-3′) were provided by Sangon Biotech Co., Ltd. (Shanghai, China). After RNA extraction was conducted using the TRIzol agent (TaKaRa, Dalian, China), the expression of the four genes in GBM and normal samples was detected using the SYBR Green Master Mix kit (Applied Biosystems, Foster City, CA). The 20 μL reaction system contained 10 μL SYBR Premix Ex Taq (2 × ), 8 μL cDNA template, 1 μL forward primer (10 μM), and 1 μL reverse primer (10 μM). The amplification conditions were as follows: 50°C for 3 min; 95°C for 3 min; 95°C for 10 s; and 60°C for 30 s for 40 cycles; Melt Curve 60°C to 95°C: Increment 0.5°C for 10 s Plate Read. GAPDH was used as the internal control gene, and all experiments had three repeats.
Statistical analysis
Based on the 2−ΔΔCt method (Arocho et al., 2006), the expression levels of the four genes were calculated. All data are shown as mean ± standard error of mean. Statistic analysis and plotting separately were performed using SPSS 22.0 (SPSS, Inc., Chicago, IL) and GraphPad Prism 5 (GraphPad Software, San Diego, CA) softwares. A p < 0.05 indicated a significant difference.
Results
DEG screening
The expression profile of five individual samples of adult neural stem cells and nine individual samples of GSCs was collected from the GEO database. By gene expression analysis, we found that a total 1130 genes were upregulated and 1296 were downregulated in GBM. Based on the mRNA-seq from the TCGA, 4971 upregulated and 4143 downregulated genes were identified in GBM. Finally, there 884 overlapping DEGs, including 418 upregulated and downregulated genes, were identified, which were used for further analyses.
GO function and KEGG pathway enrichment analyses of the DEGs
GO function and KEGG pathway enrichment analyses were conducted for the overlapping upregulated and downregulated genes. The majority of upregulated genes were mainly related to cell cycle and the downregulated genes were closely related to the transmission of nerve impulse and tight junction (Table 1).
The Top 5 Gene Ontology Function and Kyoto Encyclopedia of Genes and Genomes Pathway Enriched by the Up- and Downregulated Genes
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Construction of PPI network
The PPI network was constructed by online software STRING and visualized using the Cytoscape software. As shown in Figure 1, the network comprised a set of 3418 pairs involving 751 nodes. By analysis of topological properties of the PPI network, the score of every node was calculated and the top 10 genes, including AKT1 and CDK2, are displayed in Table 2. MCODE analysis detected seven clusters (Fig. 1) and the most significant KEGG pathways enriched by the seven clusters are listed in Table 3. Of them, genes in clusters 1 and 2 were both inventively associated with cell cycle. The genes in clusters 3, 4, 5, and 6 were mostly enriched by the pathway of cholinergic synapse, spliceosome, neuroactive ligand/receptor interaction, and extracellular matrix-receptor interaction, respectively. No pathway was significantly enriched by the genes in cluster 7.

Clusters extracted from the protein–protein interaction network with MCODE. The dark gray cycle nodes stand for upregulated genes and the light gray cycle nodes stand for downregulated genes. MCODE, Molecular Complex Detection.
The Top 10 Hub Genes in the Protein–Protein Interaction Network
The 6 Clusters with the Score >5 Reported by Molecular Complex Detection and the Kyoto Encyclopedia of Genes and Genomes Pathway That Each Is Most Enriched In
ECM, extracellular matrix; FDR, false discovery rate.
Construction of the TF-miRNA-target regulatory networks
To predict TFs targeting the DEGs, we selected TF-gene pairs based on the ITFP website and the TRANSFAC database, and finally, the TF-target network, including 52 TFs, was constructed (Fig. 2).
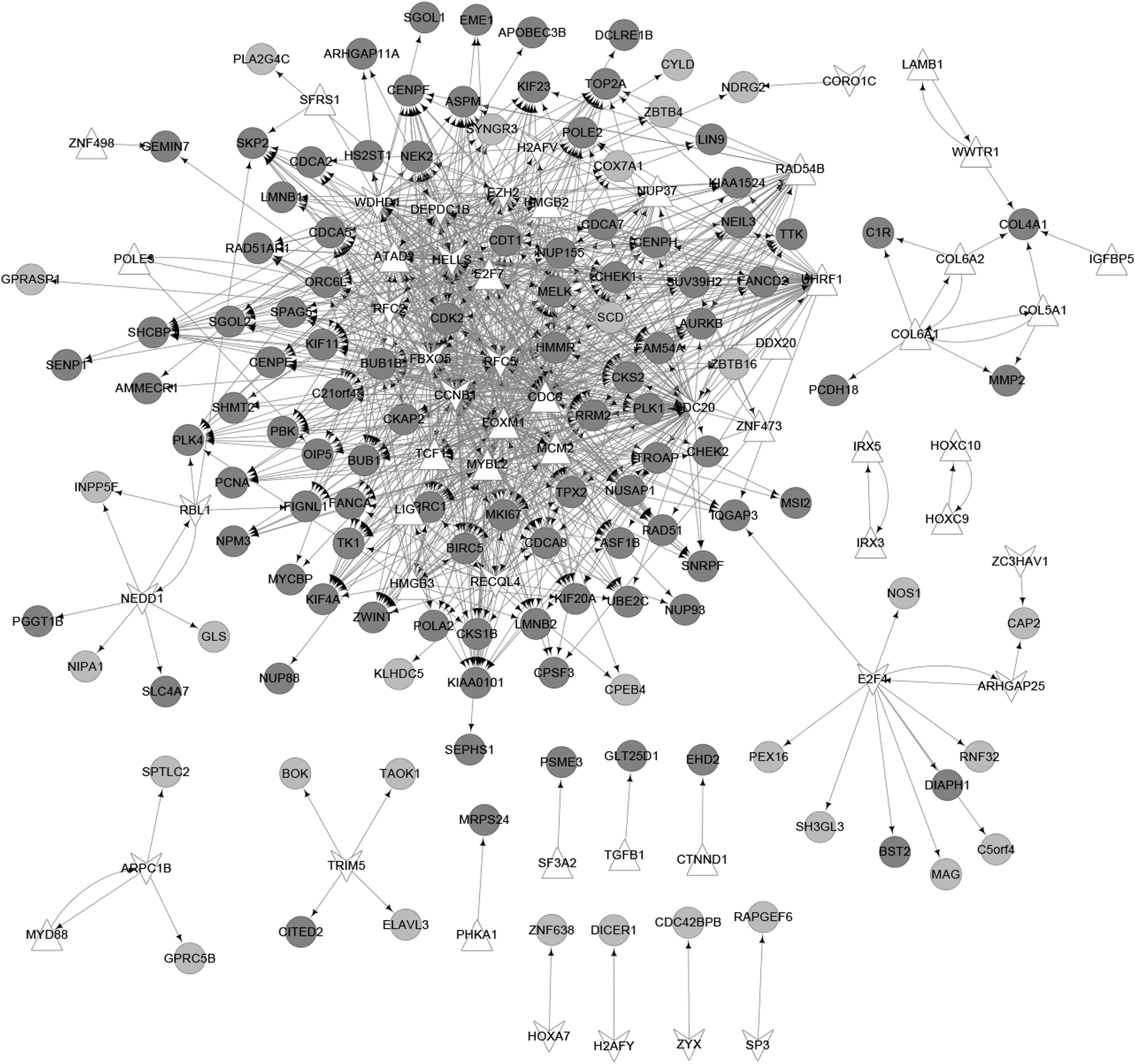
The transcriptional factors-target network. The dark gray cycle nodes stand for upregulated genes and the light gray cycle nodes stand for downregulated genes. The white regular triangle nodes and V-shaped nodes stand for up- and downregulated transcriptional factors, respectively.
Based on the miRNA-target pairs predicted using WebGestalt, an miRNA-target, consisting of 36 miRNAs and 141 genes, was constructed (Fig. 3). Next, the networks, comprising these genes and the corresponding TFs and miRNAs, were constructed (Fig. 4) by synthesizing the TF-gene pairs and miRNA-gene pairs. The top 10 miRNAs and TFs are shown in Tables 4 and 5, respectively. CCNB1 (CCNB1 cyclin B1) and RCF5 (replication factor C subunit 5) were the two TFs and miR524 and miR34B were the two miRNAs with the highest degree in the networks.
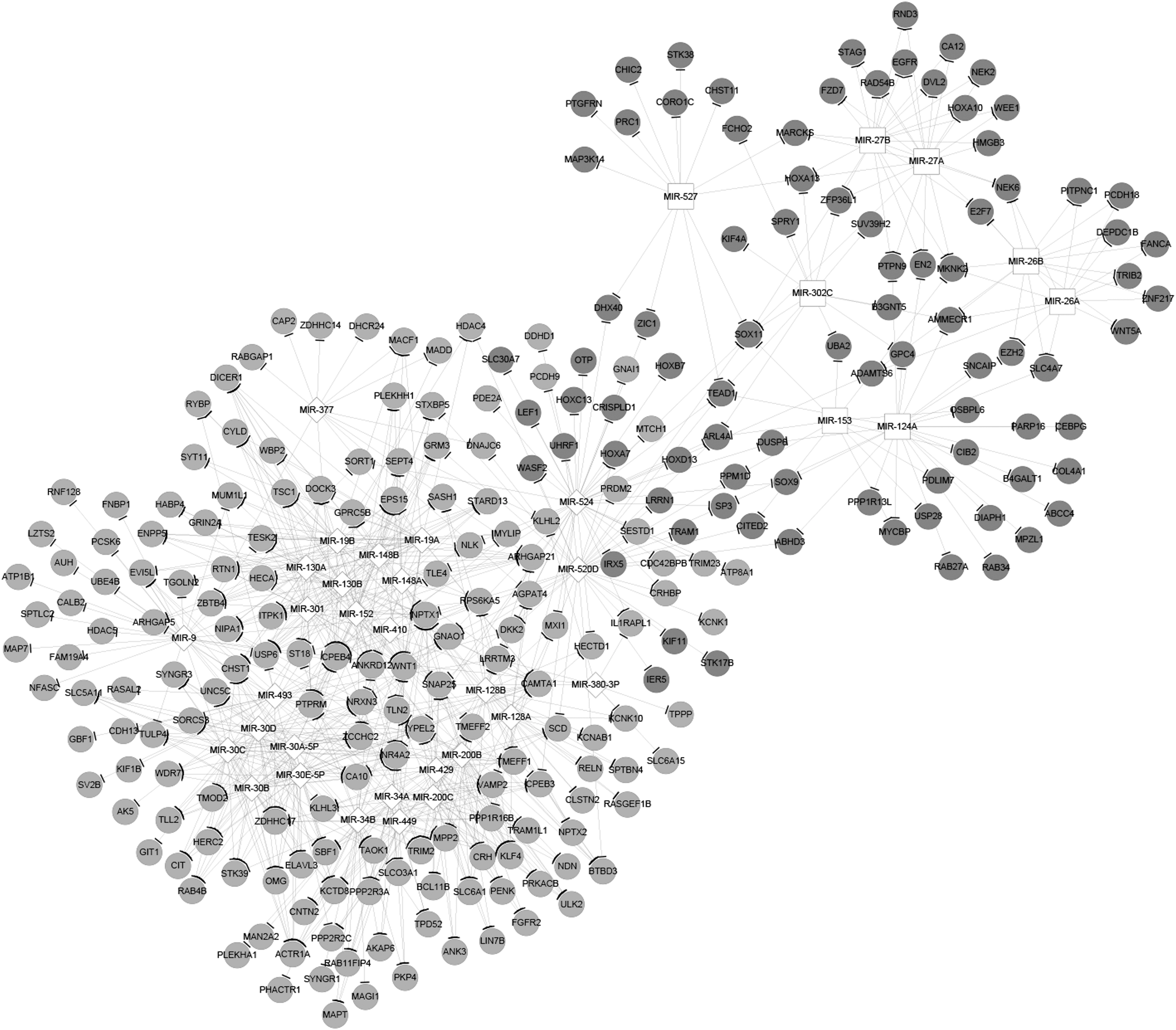
The microRNA-target network. The dark gray cycle nodes stand for upregulated genes and the light gray cycle nodes stand for downregulated genes. The white square nodes and diamond nodes stand for up- and downregulated microRNAs, respectively.

The miRNA-transcription factor-target network. The dark gray cycle nodes stand for upregulated genes and the light gray cycle nodes stand for downregulated genes. The white square nodes and diamond nodes stand for up- and downregulated miRNAs, respectively. The white regular triangle nodes and V-shaped nodes stand for up- and downregulated TFs, respectively.
The Top 10 miRNAs in the Transcriptional Factor-miRNA-Target Regulatory Network
The Top 10 Transcriptional Factors in the Transcriptional Factor-miRNA-Target Regulatory Network
Survival analysis of GBM patients
The survival curves of high- and low-expression subgroups were plotted and analyzed. There were 43 genes significantly related to the survival of GBM patients. The top 10 most relevant genes are shown in Table 6, including NPTX2 (p = 0.00097) and NRXN3 (p = 0.00068), which were negatively associated with the survival of GBM patients (Fig. 5).
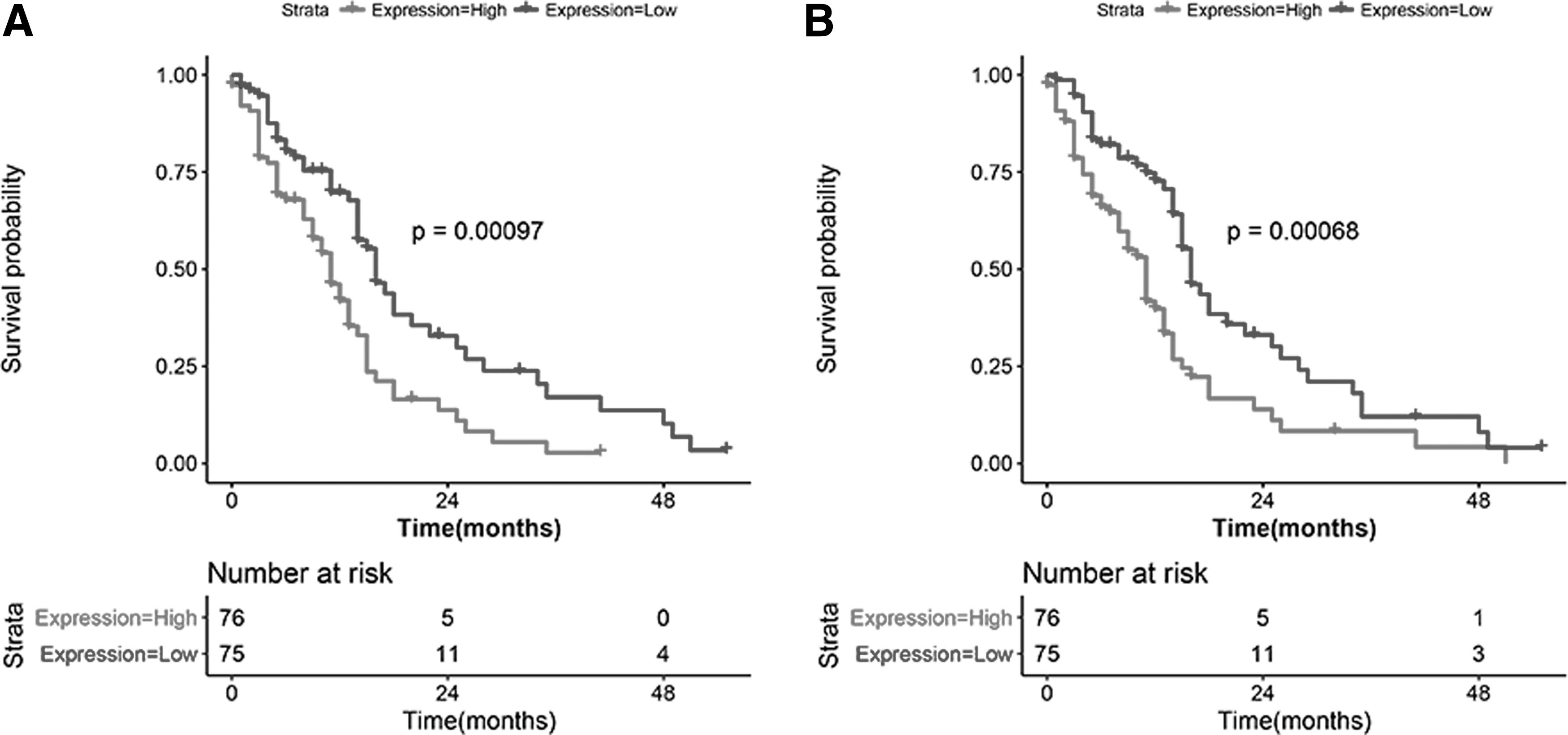
Kaplan–Meier curves for NPTX2
The Top 10 Genes Significantly Related to Survival
qRT-PCR analysis
The expression of AKT1, CDK2, NRXN3, and NPTX2 in GBM and normal samples was measured using qRT-PCR experiments. Relative to normal samples, AKT1 (p < 0.05; Fig. 6A) and CDK2 (p < 0.05; Fig. 6B) were significantly upregulated in GBM samples. Meanwhile, NRXN3 (p < 0.05; Fig. 6C) and NPTX2 (p < 0.05; Fig. 6D) in GBM samples were significantly downregulated compared with normal samples. These results were in accord with the findings of DEG screening.

The expression levels of AKT1
Discussion
GBM, characterized by invasiveness, is the most common malignant brain tumor. The prognosis of the patients is very poor following the current traditional therapeutic methods (2016; Nørøxe et al., 2017). Recently, genetic therapies for GBM have focused on improving the clinical outcomes and have achieved variable degrees of success in preclinical models and clinical trials (Kwiatkowska et al., 2013). In this study, we utilized a bioinformatic method to predict the dysregulated networks in GBM and then to identify potential biomarkers for GBM. The gene expression profile data set associated with GBM was downloaded from GEO and TCGA projects. The overlapping abnormal genes were collected for following analyses. The PPI network suggested that AKT1 and CDK2 may be the critical genes in the development of GBM. To identify the dysregulated biological processes, we conducted GO function and KEGG pathway enrichment analysis and the results showed that the most pertinent processes were cell cycle process, cholinergic synapse (related to transmission of nerve impulse), and ECM-receptor interaction. The TF-miRNA-target regulatory networks predicted that CCNB1, RCF5, miR524, and miR34B may be the critical regulators in the development of GBM. Moreover, NRXN3 and NPTX2 may be associated with the survival of GBM patients. In addition, upregulated AKT1 and CDK2, as well as downregulated NRXN3 and NPTX2, were confirmed by qRT-PCR experiments.
Cell-cycle dysregulation can lead to premature entry into mitosis, uncontrolled cell proliferation, and malignant transformation of the cell (Puyol et al., 2016). Deregulation of cell cycle controls is a cardinal feature of cancer cells (Sherr and Bartek, 2017). CDK2, CCNB1, and RCF5 were three upregulated genes and CCNB1 and RCF5 were the key TFs in the present study. They were all cell cycle regulated genes and thus they may take roles in the progression of GBM. The CDK2 protein belongs to the CDK family, which catalyzes the transfer of phosphate from ATP to specific protein substrates and thereby controls the cell cycle (Grana and Reddy, 1995). CCNB1 is also a master regulator of cell cycle progression by forming complexes with constitutively expressed CDC2 (Muschel et al., 1991). The upregulation of CDK2 and CCNB1 has been found in many types of cancers (Saxena et al., 2010; Yang et al., 2015). Besides, there was a significant correlation between the overexpression of CCNB1 and the poor outcomes in glioma patients (de Haas et al., 2008). RCF5 encodes the smallest subunit of the replication factor C complex, which is required for DNA replication. It is also one of the cell cycle-regulated genes and is most frequently upregulated during G1/S phase (Mjelle et al., 2015). Peng et al. (2017) showed that forkhead box M1 can activate the expression of RFC5 to promote temozolomide resistance in glioma cells. However, there were few studies on the implication of these genes in GBM and more studies are needed for insight into the roles of them in GBM.
AKT1, another upregulated gene in this study, encodes the RAC-alpha serine/threonine-protein kinase, which is an enzyme belonging to the AKT subfamily (AKT-1, -2, and -3) of serine/threonine kinases. The Akt/AKT1 pathway is vital for cell survival and inhibition of apoptosis, and AKT1/PKBα kinase is frequently elevated in human cancers (Sun et al., 2001). Indeed, the dysregulation of the AKT pathway was demonstrated in a majority of GBMs by large-scale genomic analysis (Riemenschneider et al., 2006; McDowell et al., 2011). The pathway leads to activated AKT and then elevated phospho-AKT levels in the majority of GBM tumor samples, leading to uncontrolled growth of tumor cells, enhancement of tumor invasion, and escape of apoptosis (McDowell et al., 2011). In parallel, AKT1 was regulated in GBM compared with the normal subjects in our study. Inhibitors of the AKT1 may be promising options for GBM treatment and additional researches are needed for fully exploring and developing this possible therapeutic strategy.
miRNAs are noncoding RNAs containing about 22 nucleotides, and regulate the expression of a wide variety of genes by directly interacting with the targeting mRNA (Bartel, 2009). miRNAs are closely involved in the initiation and progression of cancer (Garzon et al., 2009). In the present study, we constructed a TF-miRNA-target regulatory network to investigate the key regulators and found that miR524 and miR34b may be the critical miRNAs in GBM. miR34b has been reported to be related to many types of cancer, including breast cancer (Lee et al., 2011), hepatocellular carcinoma (Son et al., 2013), and gastric cancer (Suzuki et al., 2010). The miR-34 family, including miR-34a and miR-34b/c, is a direct p53 target, and the downregulation is linked to resistance against cell apoptosis (Hermeking, 2010). Little is known about the relationship between miR524 and cancer. More studies are needed to investigate the functions of miR524 and miR34b in GBM.
Furthermore, we found that NRXN3 and NPTX2 were intensively associated with the survival of GBM patients. NRXN3 and NPTX2 are two protein functions in the nervous system. GBM is the most frequent and lethal cancer originating in the central nervous system and multiple studies have reported the dysregulation of synaptic transmission and nerve impulse (Dong et al., 2010a, 2010b) in GBM. NRXN3 is one of the neurexins and takes a role in cell adhesion and recognition (Sun et al., 2013). NRXN3 is downregulated by forkhead box Q1 in gliomas and then promotes glioma cell proliferation and migration (Sun et al., 2013). NPTX2, a risky methylated gene, displayed low expression in GBMs and glioma cell lines, which could be reversed by treatment of methylation inhibitor (Shukla et al., 2013). Overexpression of NPTX2 can induce cell apoptosis, inhibit proliferation and anchorage-independent growth, and render glioma cells chemosensitive (Shukla et al., 2013). All said that CAMTA1, CPEB4, NRXN3, and NPTX2 are potential molecular targets for the prognosis or treatment of GBM.
Conclusions
In conclusion, we analyzed the DEGs between GBM and normal samples and further analyzed the dysregulated PPI network and TF-miRNA-target regulatory networks in GBM to identify the hub candidate. Moreover, the correlation between survival and abnormal genes was analyzed. Finally, AKT1, CDK2, CCNB1, RFC5, NRXN3, and NPTX2, as well as the miRNA524 and miRNA34B, were identified and they might play significant regulator roles in GBM. However, all these predicted results need to be further verified by extensive experiments.
Footnotes
Acknowledgment
This study was supported by the Natural Science Foundation of China, the National Natural Science Youth Fund Project (Grant No. 81400404).
Disclosure Statement
The authors report no conflicts of interest.