
Abstract
Trophoblast stem cells (TSCs), the precursors of placental cells, are effective for studying placental formation in vitro. Using a dual inhibition (2i) medium and mixed L-Wnt3a/mouse embryonic fibroblast feeder cells, we previously established the bovine trophoblast cell line BTS-1. In this study, we used bovine fetal fibroblasts and added Wnt3a to the 2i medium to establish another bovine TSC line (BTSW). BTSW cells expressed pluripotency markers, including NANOG, SOX2, OCT4, TRA-1-60, TRA-1-81, SSEA4, CDH1, and KRT18, and TSC markers CDX2, TEAD4, and ESRRB. Methylation sequencing of the promoter regions of NANOG, OCT4, and CDX2 revealed no significant differences between BTS-1 and BTSW cells. Removal of Wnt3a from the culture medium resulted in downregulation (p < 0.05) of NANOG, OCT4, CDX2, and TSC marker genes, and upregulation of TSC differentiation markers, including MASH2, GCM1, and PAG. Western blotting indicated activation of the WNT-YAP/TAZ signaling pathway in BTS-1 and BTSW cells, consequently activating TEAD4 transcription. However, this pathway was not activated in BCFF cells, an established bovine embryonic stem-like cell line that expresses OCT4, SOX2, and NANOG, but not CDX2. Thus, Wnt3a may play a critical role in bovine TSC maintenance by activating and regulating CDX2 expression through the WNT-YAP/TAZ signaling pathway.
Introduction
Placental nutrient and hormone availability directly affect fetal growth and development (Tanaka et al., 2001). Placental abnormalities are one of the main causes of fetal abnormalities and pregnancy failure (Godfrey, 2002). Trophoblast stem cells (TSCs) serve as precursors of the differentiated cells of the placenta. TSCs are thought to exist in all placental mammals, especially in the early stages of placental development during maximal trophoblast growth (Roberts and Fisher, 2011). TSCs can be obtained from either the trophectoderm or the extraembryonic ectoderm next to the inner cell mass (ICM) (Roberts and Fisher, 2011). Unlike embryonic stem cells (ESCs), TSCs cannot differentiate into most cell types in vivo. Nevertheless, they participate in the construction of the placenta that gradually forms after fetal trophoblast cells invade the maternal endometrium. TSCs that drive placenta formation have the ability to differentiate in vitro into various placental trophoblast cells, such as trophoblast giant cells, syncytiotrophoblasts, glycogen cells, and spongiotrophoblasts (Simmons and Cross, 2005). Therefore, TSCs can serve as a good in vitro model for the study of placenta formation.
Mouse TSCs were the first ones to be established in vitro (Tanaka et al., 1998). These TSCs were maintained under the control of fibroblast growth factor (FGF) 4 and heparin, with inactivated mouse fetal fibroblasts used as the feeder layer (Tanaka et al., 1998). Later, activin A or transforming growth factor beta 1 (TGF-β1) was used for TSC maintenance (Erlebacher et al., 2004). However, despite research efforts using the mouse (Tanaka et al., 1998; Himeno et al., 2008) and the rhesus macaque (Vandevoort et al., 2007), understanding of TSCs in other species has remained limited. Several groups have obtained different ungulate trophoblast cell lines (Ramsoondar et al., 1993; Fléchon et al., 1995; Talbot et al., 2000; Ka et al., 2001; La Bonnardiere et al., 2002; Miyazaki et al., 2002; Hashizume et al., 2006). Bovine TSCs expressing select TSC and pluripotency markers have been established using a dual inhibition (CHIR99021and PD0325901; 2i) medium and mixed L-Wnt3a/mouse embryonic fibroblast (MEF) feeder cells (Huang et al., 2014). In the placenta of cattle, there are mainly two types of trophoblast cells: mononuclear and binuclear (Wimsatt, 1951; Greenstein et al., 1958). The former accounts for a large proportion of placental trophoblast cells and is mainly responsible for nutrient exchange between maternal and fetal cells. By contrast, the binuclear trophoblast cells can synthesize many pregnancy-related hormones, such as placental lactogen (PL) and progesterone (Duello et al., 1986; Myers and Reimers, 1988), which are partly involved in pregnancy maintenance (Kazuyoshi et al., 2007).
In domestic species, including pigs (Ka et al., 2001), sheep (Hayashi et al., 2007), and cows (Degrelle et al., 2005), the trophoblast elongates dramatically before attachment, exposing a large area of the trophectoderm to the conceptuses, thus allowing them to use uterine secretions throughout the uterine lumen for growth and development (Roberts and Fisher, 2011). This dramatic period of trophoblast growth is likely sustained by the maternal system and growth factor-containing uterine secretions, rather than by the small embryonic disk (Ka et al., 2001; Spencer and Bazer, 2004).
Specific spatiotemporal expression patterns have been observed for WNT signaling pathway components during bovine placental development (Lu et al., 2013). WNT signaling plays diverse roles in mice, sheep, and humans during preimplantation events, including preimplantation embryonic development, blastocyst activation for implantation, and uterine decidualization (Tanaka et al., 1998; Hayashi et al., 2007; Sonderegger et al., 2010). Wnt3a is a classic canonical WNT ligand that plays an important role in various processes during embryonic development, including maintenance of pluripotency, cell migration during gastrulation and neurulation, and body axis formation (Denicol et al., 2013). In a recently delineated alternative WNT-YAP/TAZ signaling axis (Park et al., 2015), Wnt3a was shown to elicit both β-catenin-dependent and β-catenin-independent YAP/TAZ responses (Angers and Moon, 2009; Park et al., 2015). However, the function of WNT signaling during bovine trophoblast formation remains poorly understood.
In this study, we tested different culture systems to derive bovine trophoblast stem-like (bTS) cells or ESCs. Addition of Wnt3a to the 2i medium resulted in the establishment of the bovine TSC line termed BTSW. Removal of Wnt3a from the culture medium resulted in the downregulation (p < 0.05) of NANOG, OCT4, and CDX2, and of multiple TSC markers, together with the upregulation of TSC differentiation markers such as MASH2, GCM1, and PAG. The WNT-YAP/TAZ signaling pathway, which was activated in the TSCs, promoted the transcription of TEAD4. However, this signaling pathway was not activated in BCFF cells (a bovine ESC-like line established in this study) that expressed OCT4, SOX2, and NANOG, but not CDX2. Taken together, our data indicated that Wnt3a activates and regulates the expression of CDX2 through the WNT-YAP/TAZ signaling pathway, thereby playing a pivotal role in maintaining bovine TSC properties.
Materials and Methods
Ethics statement
All experiments with mice and cattle (generation of fetal fibroblasts) were conducted in accordance with the Guide for Care and Use of Laboratory Research Involving Animals and were approved by Inner Mongolia University's Animal Care and Use Committee.
In vitro bovine oocyte maturation, in vitro fertilization, and blastocyst acquisition
In vitro maturation of bovine oocytes, in vitro fertilization (IVF), and blastocyst harvesting were performed as previously described (Thompson et al., 2000; Oback and Wells, 2003). Bovine ovaries were obtained from a local slaughterhouse. Cumulus oocyte complexes (COCs), extracted from 3- to 8-mm follicles, were cultured for 22–24 h at 38.5°C in a humidified atmosphere with 5% CO2 in a maturation medium consisting of M199 (Gibco) supplemented with 10% fetal bovine serum (FBS; Corning), 0.05 IU/mL sheep follicle-stimulating hormone (Sigma-Aldrich), 1 μg/mL β-estradiol (Sigma-Aldrich), and 10% (v/v) penicillin-streptomycin (Gibco). Following maturation, COCs were transferred to a fertilization medium (Supplementary Table S1). After centrifugation, frozen cattle semen was mixed with the mature COCs at a final concentration of 106 sperm/mL. After 6 h, presumptive zygotes were denuded and placed in a synthetic oviduct fluid (SOF) medium (Huang et al., 2014) containing 0.6% bovine serum albumin (BSA; Sigma-Aldrich). After 48 h, cleaved embryos were transferred to fresh SOF medium with 5% FBS. Following 7 or 8 days of culturing, fully expanded blastocysts were collected for subsequent experiments.
Feeder cell preparation
L-Wnt3a cells (CRL-2647™, ATCC®) were cultured in Dulbecco's modified Eagle medium (DMEM)/F12 (Gibco) supplemented with 10% (v/v) FBS for expansion. MEFs were derived from E13.5 embryos. Primary cells, prepared by mincing fetal tissue after head, heart, and liver removal, were cultured using 100-mm dishes in high-glucose DMEM (Gibco) supplemented with 10% FBS (Corning). Monolayer cells, at ∼70% confluence, were harvested by trypsinization (0.05% trypsin; Gibco). This was considered passage 1. L-Wnt3a cells and MEFs, mixed at a ratio of 1:1, were treated with 17 μg/mL mitomycin C (Sigma-Aldrich) for 2.5–3 h at 37°C. The cells were then washed thrice with Dulbecco's phosphate-buffered saline (DPBS; BI) and frozen in FBS with 10% dimethyl sulfoxide (Sigma-Aldrich) as hybrid feeder cells termed L-Wnt3a cells/MEFs (L/M).
Bovine fetal fibroblasts (BFFs) were derived from bovine fetuses obtained from the same slaughterhouse as above. BFF primary cells were cultured from fetal bovine ear tissues in 100-mm dishes using high-glucose DMEM supplemented with 10% (v/v) FBS. The BFFs were trypsinized, treated, and frozen to prepare feeder cells as described above. Feeder cells were seeded at 105 cells/well in four-well culture plates (Nunc, Denmark) before subculture of the bovine TSCs.
Derivation of bTS cells and ESC-like cells
IVF-derived bovine blastocysts (day 7 and 8) were used to obtain bTS and ESC-like cells, which were transferred onto the feeder cells after removal of the zona pellucida using pronase (2 mg/mL, Roche). After outgrowth formation, the cells were mechanically collected and seeded into a new well using a syringe needle. The medium was changed daily, and the cells were subcultured onto fresh feeder cells weekly. The 2i medium consisted of DMEM/F12 and Neurobasal™ Medium (Gibco) with PD0325901 (0.5 μM; Selleckchem), CHIR99021 (3 μM; Selleckchem), B-27™ Supplement (10 mL/L; Gibco), N-2™ Supplement (5 mL/L; Gibco), and Glutamax™ Supplement (10 mL/L; Gibco) (Li et al., 2008). When indicated, recombinant human Wnt3a (100 ng/mL; R&D Systems) was added to the medium for cell culture. The derived bTS cells were named BTSW.
To obtain ESC-like cells, the feeder cells were replaced with Matrigel (10 μL/well; Corning). Cells were grown in CHIR99021 medium, corresponding to the 2i medium without PD0325901, and supplemented with 50 ng/mL recombinant human basic FGF (bFGF; Promega) and 10 μmol/L forskolin (Sigma-Aldrich), which was named as CHIR99021, bFGF, Forskolin (CFF) medium. The derived cells were named BCFF. Following incubation of the blastocysts in CFF medium for 3–4 days, the ICM was separated from trophectoderm by mechanical cutting using a syringe needle and transferred to a new well to be cultured under the same foster conditions as described above. Following cell colony formation, the cells were collected and seeded in the same way as the bTS cells. The medium was changed daily, and the cells were passaged onto fresh Matrigel weekly.
Immunofluorescent staining
Cells were washed once in DPBS and fixed in 4% paraformaldehyde (Solarbio, China) for 15 min. Following a 20-min permeabilization with 0.5% (v/v) Triton X-100 (Solarbio) in DPBS, the cells were blocked for 1 h at room temperature using 10% (v/v) goat serum in DPBS (blocking solution). The samples were then incubated overnight at 4°C with primary antibodies (Supplementary Table S2) diluted in the blocking solution. Following three washes, samples were incubated for 1 h at room temperature with secondary antibodies (Supplementary Table S2) diluted in the blocking solution. Following two washes, the cell samples were stained for 5 min at room temperature with 4′,6-diamidino-2-phenylindole (0.5 μg/mL; Beyotime, China) diluted in DPBS. After two DPBS washes, the cells were loaded onto slides with antifade solution (Solarbio). Negative control samples were prepared using the same procedure in the absence of primary antibodies. Nikon confocal laser-scanning microscopes (Nikon, A1) were used to visualize the fluorescent signals. Images were analyzed using the FV10-ASW 2.1 Viewer software. Image acquisition, analysis, and processing were standardized for each experiment.
Alkaline phosphatase staining
Two distinct methods were used for alkaline phosphatase (AP) staining of bTS cells and BCFF cells. Before AP staining, bTS cells were cultured on feeder cells for 3–4 days. Following the removal of the 2i medium, cells were washed with DPBS and fixed for 2 min at room temperature using 4% paraformaldehyde. Fixed cells were washed thrice with DPBS and stained for 30–40 min at room temperature in the dark using the 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium chloride Color Development Substrate Kit (Promega). Cells were washed with DPBS to terminate the staining reaction and were subsequently maintained in DPBS. The staining results were examined under a microscope (Nikon, Eclipse Ti-S).
BCFF cells were cultured on Matrigel for 2–3 days before AP staining. Following the removal of the CHIR99021 medium, cells were washed twice with DMEM/F12 and stained for 30 min at 37°C using the TransDetect AP Live Kit (TransGen Biotech, China). After removal of the AP staining solution, cells were washed twice with DMEM/F12 and maintained in DMEM/F12 for subsequent observation. The staining results were examined using an inverted fluorescence microscope (Nikon, Eclipse Ti-S).
Reverse transcription polymerase chain reaction and real-time quantitative polymerase chain reaction
Following cell lysis using the Trizol Reagent (Takara, Japan), total RNA was extracted using the TaKaRa MiniBEST Universal RNA Extraction Kit (Takara) according to the manufacturer's protocol. RNA quality and quantity were determined using a NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific). Reverse transcription and cDNA synthesis were performed using 1 μg of total RNA and the PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara). Reverse transcription polymerase chain reaction (RT-PCR) amplification was performed on an Applied Biosystems ThermoCycler (Life Technologies) with the EasyTaq PCR Supermix (TransGen Biotech) using the following protocol: denaturation at 95°C for 1 min; 35 cycles of 95°C for 30 s, 59–60°C for 30 s, and 72°C for 35 s; and final extension of 72°C for 10 min). PCR products were separated on 1.5% (w/v) agarose gels and visualized using GelStain (TransGen Biotech).
Quantitative PCR (qPCR) amplification was conducted using an ABI 7500 Real-Time PCR System (Life Technologies). Each qPCR reaction mixture contained 2 μL cDNA, 0.8 μL of each primer (0.4 μM), and 10 μL SYBR Premix Ex Taq™ II (Takara) in a total volume of 20 μL. All reactions were performed in triplicate, and product identity was confirmed by gel electrophoresis and melting curve analysis. The relative expression levels were determined using the 2−ΔΔCT method and normalized against GAPDH levels. The primer sequences used for RT-PCR and qPCR are listed in Supplementary Tables S3 and S4.
Methylation analysis
BTS-1 (p147, p204, and p209), BTS-2 (p25, p42, and p51), and BTSW (p31, p50, and p99) cells were used to analyze the promoter methylation of OCT4, NANOG, SOX2, and CDX2 by bisulfite-sequencing PCR. DNA treatment and methylation-specific PCR were executed using the ZYMO EZ DNA Methylation-Gold Kit (ZYMO RESEARCH) and Takara Ex Taq® (Hot Start Version) according to the associated manufacturer's protocols. The PCR products were ligated into pEASY ®-T1 Cloning Vector (TransGen Biotech) for methylation sequencing. At least 10 clones per gene were sequenced and analyzed for each sample. The primers used for amplification of the methylation regions of the four genes are provided in Supplementary Table S5.
Western blotting
Cell samples were washed twice with DPBS and collected using the Mammalian Protein Extraction Reagent (CWBIO, China). Sample protein concentrations were determined using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Protein samples (15 μg) combined with SDS-PAGE loading buffer (CWBIO) were loaded onto 8% or 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels. Following separation, the proteins were transferred onto 0.22-μm nitrocellulose membranes (Millipore) at 100 V for 30–100 min (depending on the study) at 4–8°C. After transfer, membranes were incubated for 1 h at room temperature in a blocking buffer (Tris-buffered saline, pH 7.4 with 0.1% [v/v] Tween 20 and 5% [w/v] BSA). The membranes were then incubated at 4°C overnight with primary antibodies (Supplementary Table S6) diluted in the blocking buffer on an orbital shaker. After washing, the membranes were incubated for 1 h at room temperature with secondary antibodies (Supplementary Table S3) diluted in the blocking buffer. Immunoreactive proteins were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), and images were obtained using a Tanon 5200 Multi Automatic Fluorescence and Chemiluminescence Imaging System (Tanon, China). Stripping Buffer (CWBIO) was used according to the manufacturer's instructions to detect the total protein and phospho-specific bands on the same blots.
Statistical analysis
Statistical analyses of qPCR and DNA methylation results were performed using the SPSS 20 software (IBM), and statistically significant differences between groups were identified using a Student's t-test. p-Value less than 0.05 was considered statistically significant.
Results
bTS cell lines can be established in a 2i medium in the presence of Wnt3a
The bovine blastocyst formation rate from four IVF experiments was determined to be 26.4% (144/544, Supplementary Table S7). Following zona pellucida removal, 91 blastocysts, selected on day 7–8, were cultured using different culture systems (Supplementary Table S8). Although some blastocysts attached to the feeder cells after 3–4 days, most cells remained floating, and the cavity space continued to increase. The culture system with a feeder layer demonstrated a higher adherence rate than that without feeder cells. After 6–7 days of cultivation, the adherent blastocysts formed outgrowths in the 2i medium with the L/M or BFFs as feeder cells (Supplementary Fig. S1A, B), but not in the CHIR99021 medium without feeder cells (data not shown).
Two types of 2i systems were used to obtain bTS cells: the 2i+L/M system comprised the 2i medium and L/M feeder cells, whereas the 2i+Wnt3a-BFF system comprised the 2i medium supplemented with exogenous Wnt3a (100 ng/mL) and BFF feeder cells. The obtained cells had typical morphology of mouse TSCs and could be cultured in vitro long term (Supplementary Fig. S1A, B). Three TSC lines were established from the bovine IVF blastocysts. The BTS-1 line, previously identified as bovine TSCs (Huang et al., 2014), had been cultured for more than 200 generations over 48 months (Supplementary Fig. S1B). Two additional lines, which we named BTS-2 and BTS-3, were passaged for over 50 and 150 generations, respectively (Supplementary Fig. S1B). Using nuclear transfer blastocysts, we also obtained two bTS cell lines, termed as BTSA and BTSB, both of which were cultivated for more than 100 generations in vitro. The four cell lines were similar in morphology to the BTS-1 cells, characterized by dome structures and suspension of vesicles.
Using the 2i+Wnt3a-BFF system, we obtained two cell lines, named BTSE (data not shown) and BTSW (Supplementary Fig. S1B). These cells, which were passaged in vitro for more than 20 and 120 generations, respectively, exhibited morphology very similar to that of BTS-1 cells. The CHIR99021 medium, supplemented with bFGF and forskolin, facilitates the establishment of bovine ESC-like cells. The established BCFF cell line was used as control for further experiments (Supplementary Fig. S2).
Properties of the BTSW bTS cells are maintained by Wnt3a
As the BTSW cells displayed a stable morphology upon in vitro passaging for over 120 generations, we used RT-PCR and immunofluorescence staining to assess TSC markers in these cells. The BTSW cells were positive for the pluripotency marker OCT4 (Fig. 1A, C) and the TSC marker CDX2 (Fig. 1C), with both markers localized in the nuclei, which is consistent with the staining results seen in blastocysts (Supplementary Fig. S3; Supplementary Videos S1–S4) as well as with the staining patterns of SOX2 and NANOG. In contrast, NANOG protein levels were almost undetectable in BTS-1 cells (Huang et al., 2014). Furthermore, positive immunofluorescent staining results were also obtained for other pluripotency and TSC markers, including SSEA4, TRA-1-60, TRA-1-81, CDH1 (E-CADHERIN), and KRT18 (Fig. 1A, B). RT-PCR analysis, using BTS-1 and BTS-2 cells as controls, demonstrated the expression patterns of TSC and trophoblast markers, including CDX2, TEAD4, ELF5, GATA3, SMARCA4 (BRG1), ETS2, ESRRB, CDH3, EOMES, GRN, and IFNT2, in BTSW cells (Fig. 1E). In addition, BTSW cells were positive for AP activity, whereas the BFF feeder cells were negative for AP staining (Fig. 1D).
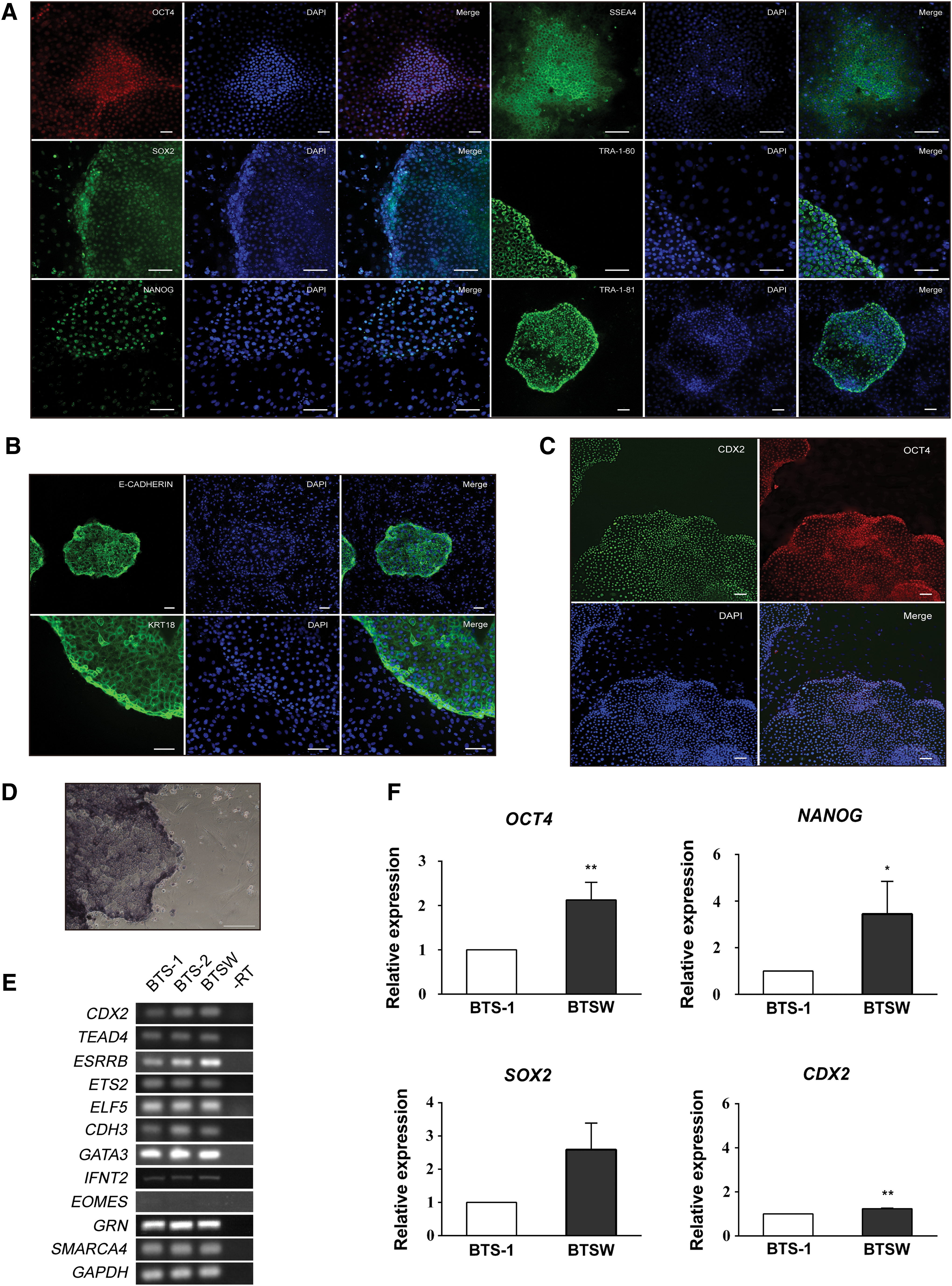
BTSW cells express pluripotency and TSC markers.
To further verify the pluripotent state of BTSW cells, we performed qPCR analysis of pluripotency genes OCT4, SOX2, and NANOG and of the TSC-specific gene CDX2 in the BTSW and BTS-1 cell lines. Expression of CDX2, OCT4, and NANOG was significantly higher in BTSW cells relative to BTS-1 cells, whereas SOX2 mRNA levels were slightly higher in BTS-1 cells (Fig. 1F). These results indicated that, despite lack of colony morphology differences between the BTSW and BTS-1 lines, the pluripotency state of BTSW cells was significantly stronger compared with BTS-1 cells. Furthermore, the BTSW cells established using the new 2i+Wnt3a-BFF system may represent an intermediate state between ESCs and TSCs.
To verify the in vitro differentiation potential of BTSW cells, especially the ability to differentiate into secretory binuclear trophoblast cells, we cultured BTSW cells in a feeder-free differentiation medium (high-glucose DMEM supplemented with 10% [v/v] FBS) for 7 days. Our results showed that the characteristic morphology of TSC colonies soon disappeared and the cells gradually become larger and flatter (Fig. 2Aa, b). By the fourth day of culture, a certain number of morphologically distinct binuclear cells had been formed in the colonies (Fig. 2Ac, d), and these cells were positive for PL immunofluorescence staining; however, this staining was not observed in undifferentiated BTSW cells (Fig. 2B). These results indicated that BTSW cells were able to differentiate into mature placental trophoblast cells in vitro.
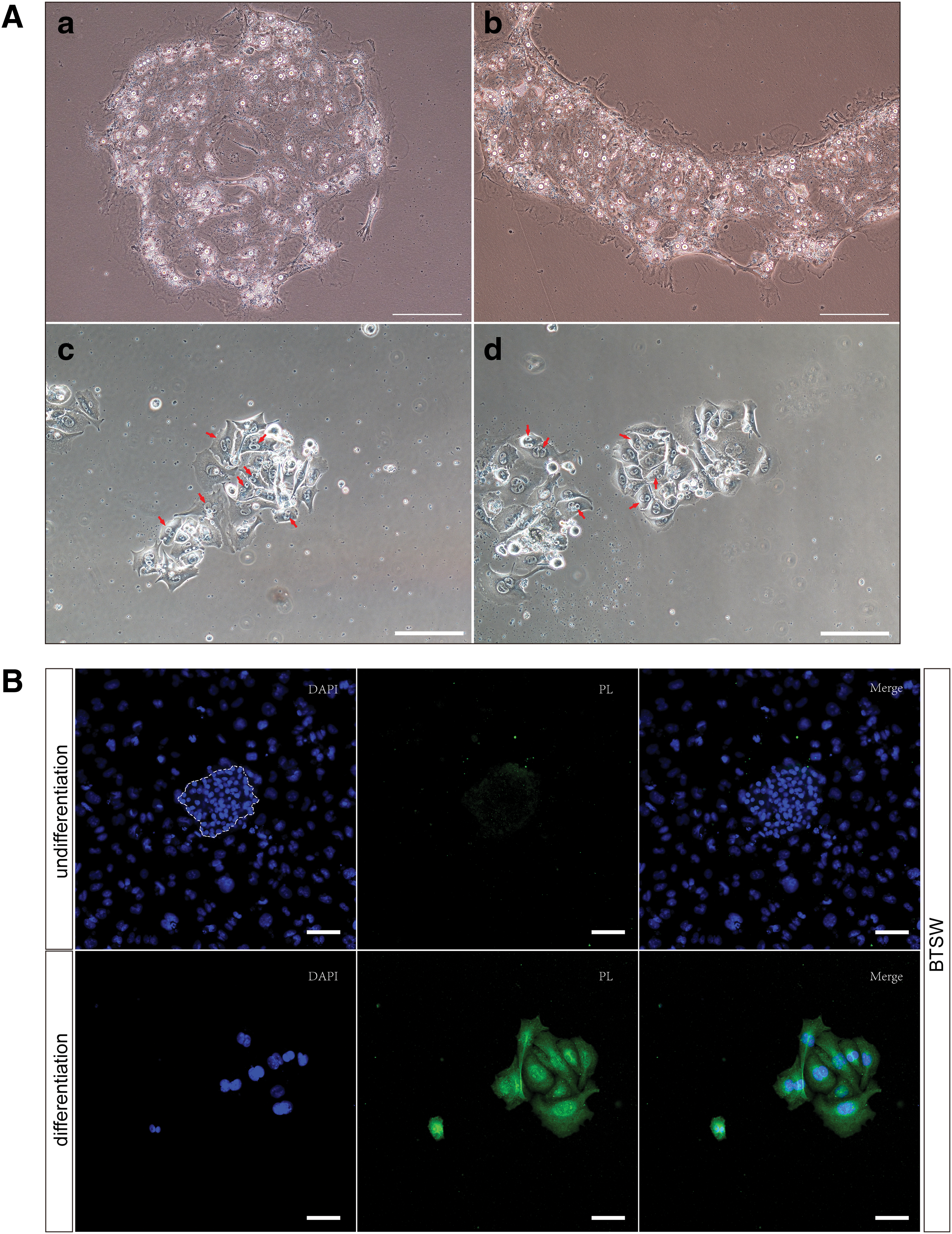
BTSW cells differentiate into binuclear cells and synthesize PL.
The results indicated that the 2i-L/M system previously used to culture bovine TSCs can be substituted with a more stable and controllable system involving 2i medium supplementation, with recombinant Wnt3a using BFFs as the feeder layer. Our findings also suggested that Wnt3a plays an important role in the maintenance of bovine TSCs.
Wnt3a maintains the self-renewal capacity of mouse ESCs. To assess whether Wnt3a has a similar effect on bovine TSCs, we performed qPCR analysis of BTSW cells (p109) cultured for 7 days using the 2i-BFF or the 2i+Wnt3a-BFF system. In contrast to the 2i+Wnt3a-BFF system, the 2i-BFF system was characterized by gradual flattening of the cells on the edge of the colonies and by an increase in cell volume following 7 days of incubation (Fig. 3A). Levels of NANOG and OCT4 mRNAs decreased by 26% and 73% (p < 0.05), respectively, in the absence of Wnt3a. Abundance of CDX2 mRNA, the central TSC marker, declined by 46% (p < 0.01) in the absence of Wnt3a. No significant effect on SOX2 transcript abundance was observed (Fig. 3B). Furthermore, almost all TSC marker genes tested using qPCR, including EOMES, ESRRB, GATA3, ETS2, ELF5, SMARCA4, and KRT18, demonstrated downregulation in the absence of Wnt3a (Fig. 3C). Transcript abundance of CDH3, however, was similar in the two groups (Fig. 3C). We also assessed the effect of Wnt3a on several major TSC differentiation marker genes, including CDH3, HAND1, MASH2, PAG, GCM1, PL-I, and GRN. As revealed by qPCR analysis, Wnt3a removal resulted in 2.36-, 1.31-, 0.36-, and 0.12-fold increases in the abundance of MASH2, PAG, GCM1, and PL-I transcripts, respectively, but had no effect on GRN mRNA levels. In contrast, HAND1 mRNA abundance increased by 77% in the absence of Wnt3a (Fig. 3D). Despite differences in expression patterns of individual genes, the above results indicated that Wnt3a not only maintained TSC pluripotency but also promoted the expression of specific TSC marker genes and maintained TSC characteristics.
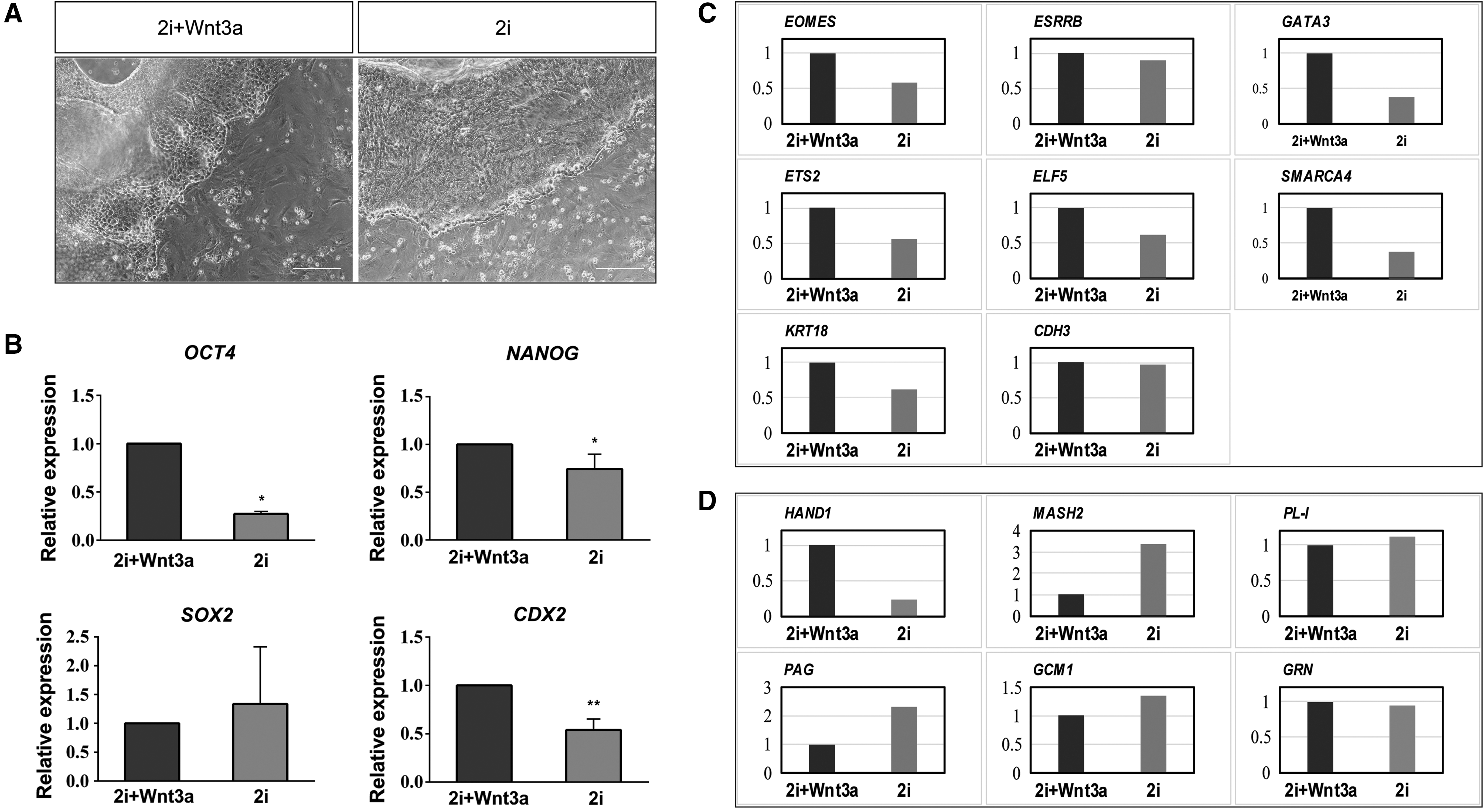
Wnt3a increases the expression of pluripotency and TSC markers in BTSW cells.
Reduced promoter methylation stimulates expression of OCT4, NANOG, and CDX2 in BTSW cells
DNA methylation is a major epigenetic modification, which preferentially occurs at the 5′ position of the cytosine in CpG dinucleotides and in clusters known as CpG islands (Gardiner-Garden and Frommer, 1987). Gene promoter methylation, which is generally thought to lead to transcriptional silencing (Larsen et al., 1992; Plass and Soloway, 2002), plays an important regulatory role in gene expression. In the absence of other factors, DNA methylation negatively correlates with gene expression.
To investigate whether the observed differences in OCT4, NONOG, and CDX2 expression were driven by altered promoter methylation patterns, we used bisulfite-sequencing PCR to compare methylation levels of the respective promoter regions in BTSW, BTS-1, and BTS-2 cells. The BTSW cell line demonstrated the lowest degree of promoter methylation for all genes tested (Fig. 4 and Supplementary Table S9). Consistent with the qPCR results that showed higher NANOG expression in BTSW cells than in BTS-1 cells (Fig. 2F), the cell lines exhibited differences in methylation frequency distribution at the NANOG promoter (Supplementary Fig. S4). These differences in methylation patterns may account for the NANOG protein being detectable in BTSW cells, but not in BTS-1 cells. The above results suggested that BTSW cells may maintain a more primitive pluripotent state than the TSC line obtained in the previous 2i-L/M culture system.
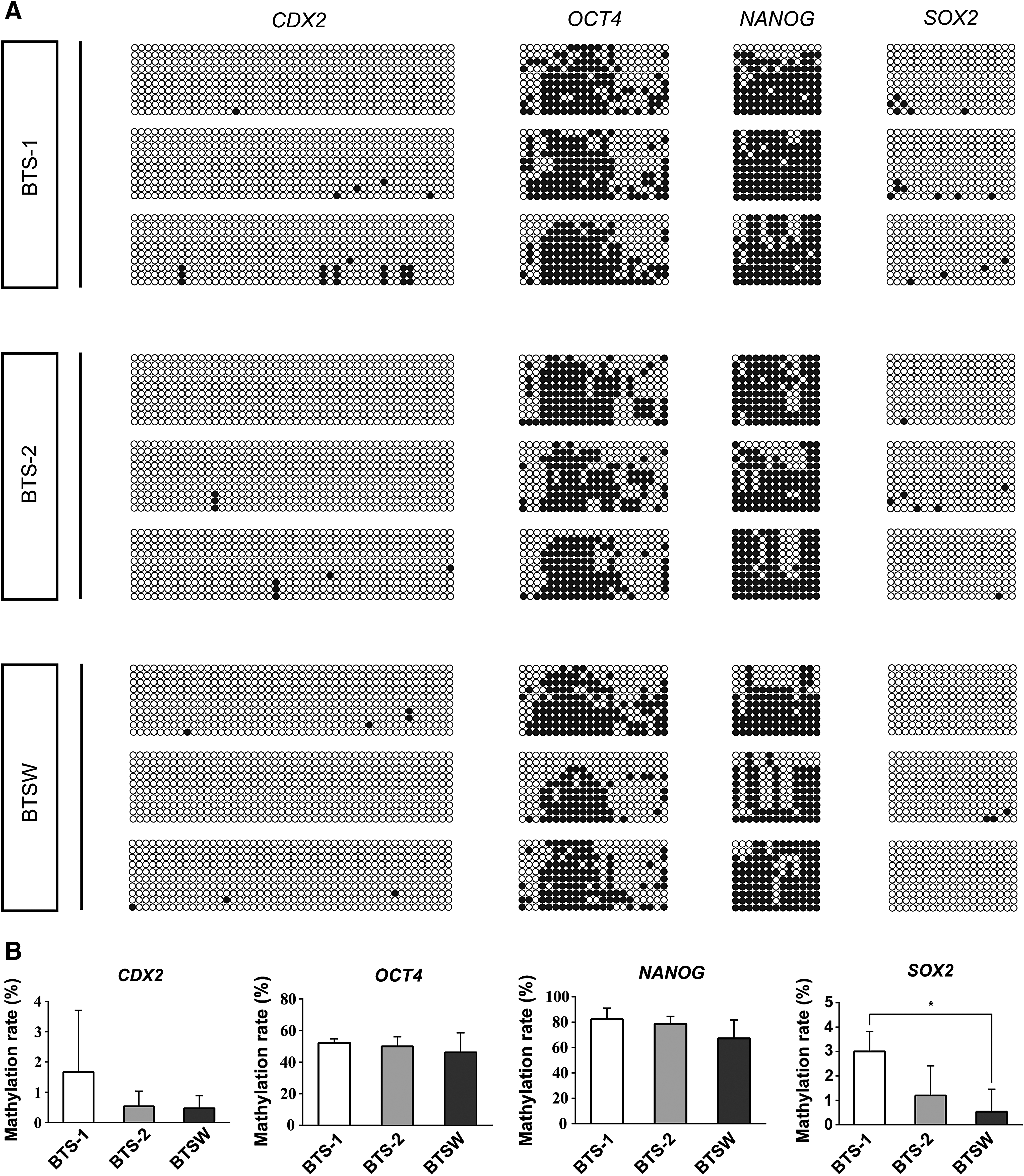
CDX2, NANOG, OCT4, and SOX2 promoters exhibit lower methylation levels in BTSW than in BTS-1 or BTS-2 cells.
Wnt3a activates the WNT-YAP/TAZ signaling pathway to sustain CDX2 expression in bTS cells
An alternative Wnt3a-activated WNT-YAP/TAZ signaling axis has been reported in the mouse, consisting of WNT-FZD/ROR-Ga12/13-Rho-Lats1/2-YAP/TAZ-TEAD (Park et al., 2015). YAP/TAZ acts as a downstream core regulator of this signaling pathway, binding to TEAD to form a transcription complex that initiates the transcription of the key TSC marker CDX2 (Supplementary Fig. S5). Hence, we hypothesized that the bovine WNT-YAP/TAZ signaling axis functions similarly. Production of several key members of the WNT-YAP/TAZ signaling pathway, including FZD1, RHOA, GNA12, LATS1, YAP, TAZ, and TEAD4, was assessed by western blotting in BTS-1, BTSA, BTSW, and BCFF cell lines. L/M cells were used as controls. Although detected in all three TSC lines, the selected proteins were activated more strongly in the BTS-1 and BTSA cells than in the BTSW cells (Fig. 5A, B), potentially due to different Wnt3a concentrations in the culture systems. In contrast, despite the production of some proteins such as FZD1, Gα12, RHOA, and TAZ, the most critical downstream transcriptional factor of the pathway, YAP, was not detectable in BCFF cells (Fig. 5B). The inactivated state of the WNT-YAP/TAZ signaling pathway in BCFF cells was further underscored by the lack of detectable TEAD4, which was present in all TSC lines (Figs. 2E and 5B, D). Consistently, expression of the TEAD4 target gene, CDX2, was detected in the BTS-1 and BTSW lines, but not in BCFF cells (Fig. 5D). These findings underscored the lack of CDX2 in BCFF cells, as revealed by immunofluorescence staining.
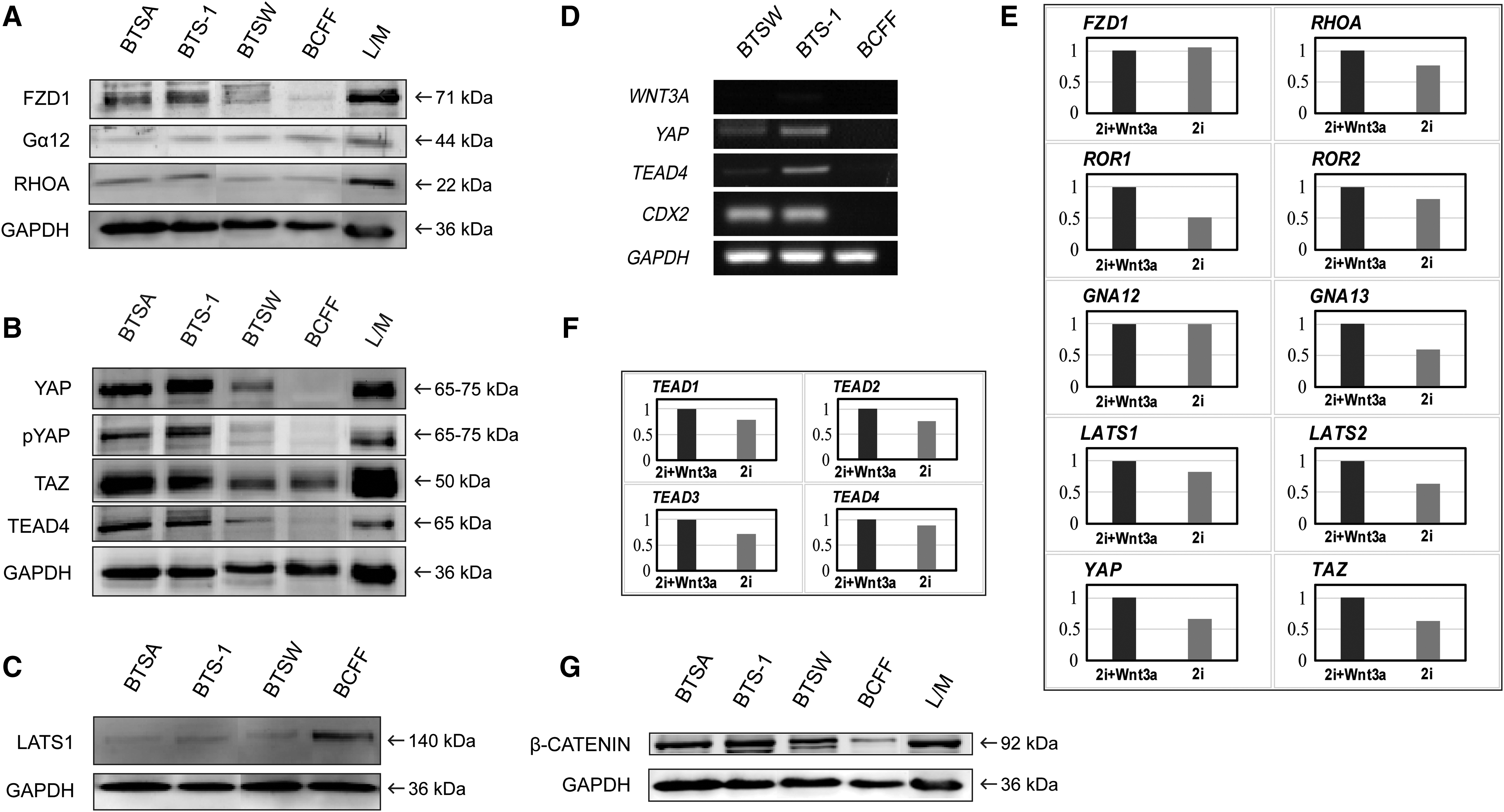
Wnt3a is an activator of WNT-YAP/TAZ signaling in TSCs.
LATS1 inhibits YAP/TAZ accumulation in the cytoplasm and transport to the nucleus by phosphorylation of YAP. When WNT-YAP/TAZ signaling is activated, RHOA inhibits LATS1 expression, thereby promoting YAP/TAZ function (Park et al., 2015). Consistent with these findings, we observed that LATS1 was present at lower levels in the three bovine TSC lines relative to the BCFF line (Fig. 5C).
To further assess whether Wnt3a indeed affects WNT-YAP/TAZ signaling, we cultured the BTSW cells for 7 days using the 2i-BFF and 2i+Wnt3a-BFF systems. Most members of the WNT-YAP/TAZ pathway, including RHOA, ROR1/2, GNA13, LATS1/2, YAP, and TAZ, as well as TEADs, which are targets of WNT-YAP/TAZ signaling (Park et al., 2015), were downregulated at the transcriptional level in the absence of Wnt3a (Fig. 5E, F), suggesting that Wnt3a affects WNT-YAP/TAZ signaling. These results indicated that the WNT-YAP/TAZ pathway is activated in bovine TSCs, and the signaling is modulated by Wnt3a in a dose-dependent manner. These findings also indicated that Wnt3a can promote the establishment of bovine TSCs by activating WNT-YAP/TAZ signaling and thereby regulating and maintaining CDX2 expression.
In addition, because Wnt3a is a ligand for both the WNT-YAP/TAZ and WNT/β-catenin signaling pathways, we also examined the β-catenin protein level, which represents activation of the WNT/β-catenin pathway. This level was quantified in bTS-1, BTSA, and BCFF cells, with L/M cells used as positive control. The β-catenin protein level was higher in the two bTS cell lines cultured in the 2i-L/M system than that in BTSW cells cultured in the 2i+Wnt3a/BFF system, whereas the level was lowest in BCFF cells (Fig. 5G). This indicated that WNT/β-catenin signaling also contributed to the proliferation and self-renewal of the bovine TSCs and suggested that the concentration of Wnt3a in the 2i-L/M system may be higher than that in the 2i+Wnt3a/BFF system. The detection of β-catenin protein in BCFF cells also suggested that the canonical WNT signaling pathway might play an important role in maintaining the self-renewal of bovine ESCs.
Discussion
The derivation and self-renewal of ESCs and TSCs require different inputs. For example, while mouse ESCs depend on LIF/STAT3 signaling, mouse TSCs require FGF4 signaling (Tanaka et al., 1998) and components released by feeder cells, such as TGF-β and activin (Erlebacher et al., 2004), to sustain proliferation in an undifferentiated state. Yet other studies have shown the establishment and maintenance of TSCs from rhesus monkeys (Vandevoort et al., 2007), voles (Grigor'eva et al., 2009), and other species in the absence of FGF4, suggesting species-specific growth factor requirements. In 2017, extended pluripotent stem cells (EPSCs), which possess embryonic and extraembryonic potency, were established from early embryos or generated from pluripotent stem cells (PSCs) by addition of small molecular compounds and cytokines to the culture system (Yang et al., 2017a, 2017b; Bao et al., 2018). The bona fide TSC lines and extraembryonic endoderm stem cells can be directly derived from EPSCs in vitro (Yang et al., 2017a). These findings suggested the feasibility of generating stable stem cell lines that have both embryonic and extraembryonic potency. The dual inhibition 2i culture system, containing CHIR99021 and PD0325901, was originally reported for the in vitro establishment and culturing of ESCs (Li et al., 2008), but this system is able to promote and maintain the expression of CDX2 in rat ESCs, thus rendering these cells the potential to differentiate into trophoblasts (Meek et al., 2013). The 2i system is unable to maintain proliferation of bovine ESCs (Eliza et al., 2015), and the bovine ESC-like cells (BCFF cells) that we established also failed to maintain a stable mouse ESC-like colony morphology in 2i medium (Supplementary Fig. S1C). Using the modified 2i-L/M and 2i-BFF systems with the addition of Wnt3a, we have established several bovine blastocyst-derived cell lines, which not only exhibited characteristics of TSCs but also showed expression of pluripotent markers. However, these cells were more similar in nature to TSCs, rather than to ESCs. Pluripotent marker genes such as NANOG and SOX2 were expressed at very low levels in these cells. The inhibition of GSK-3β by CHIR99021 may render these cells with ESC-like properties. On the other hand, these cells are double positive for OCT4 and CDX2 expression, which resemble the properties of the trophectoderm of bovine blastocysts (Supplementary Fig. S3). These characteristics distinguish ungulate embryos from rodent embryos (Ozawa et al., 2012). Flow cytometry analysis showed that over 90% of BTSW cells were double-positive for OCT4 and CDX2, which indicated that they were bipotential cells rather than a mixed cell line.
The WNT/β-catenin signaling pathway plays an important role in maintaining and promoting ESC self-renewal (Hao et al., 2006; Anton et al., 2007; Miyabayashi et al., 2007; ten Berge et al., 2011). However, WNT/β-catenin signaling can also promote trophectoderm differentiation during early embryo development (Denicol et al., 2013; Krivega et al., 2015) and differentiation of ESCs (Anton et al., 2007; Davidson et al., 2012). Overexpression of β-catenin in mouse TSCs leads to their transition to syncytiotrophoblast layer II cells. (Zhu et al., 2017). Similarly, high-level expression of CDX2 stimulated by Wnt3a promotes differentiation of mouse ESCs into TSCs (He et al., 2008). We found that NANOG, OCT4, CDX2, and multiple TSC marker genes were downregulated, whereas a subset of TSC differentiation marker genes (e.g., MASH2, GCM1, and PAG) were upregulated, in the absence of Wnt3a. We propose that low Wnt3a concentrations correspond with low-level activation of WNT/β-catenin signaling, which facilitates and maintains the expression of pluripotency-related genes. In contrast, a high level of WNT signaling activation likely leads to the overexpression of certain trophoblast cell markers, thereby disrupting the balance between TSC and pluripotency gene expression patterns, eventually resulting in irreversible differentiation of TSCs. Therefore, we propose that activation of low-level WNT-β-catenin signaling is indispensable for maintaining the stemness of bovine TSCs.
We also used the CHIR99021 cultivation system supplemented with bFGF and forskolin to establish the BCFF cell line, which was derived from the ICM isolated from bovine IVF-derived blastocysts. The appearance of these cells most closely resembled that of mouse naive-state PSCs. Previous studies have shown that the activation of the WNT/β-catenin pathway can inhibit transition from the naive to the primed state of mouse and human ESCs (ten Berge et al., 2011; Xu et al., 2016). However, a recent report showed that suppression of WNT/β-catenin signaling is required for the establishment of bovine primed pluripotent ESCs (Bogliotti et al., 2018). These studies indicated that the WNT/β-catenin signaling pathway plays an important role in the establishment of ESCs, with fine-tuning of this pathway giving rise to different outputs.
Although low-level WNT/β-catenin signaling is necessary for the maintenance of bovine TSC stemness, self-renewal of bTS cells depends to a greater extent on the WNT-YAP/TAZ signaling pathway. The WNT-YAP/TAZ signaling axis consists of WNT-FZD/ROR-Gα12/13-Rho-Lats1/2-YAP/TAZ-TEAD (Park et al., 2015). We showed that all members of the WNT-YAP/TAZ pathway were present in the three TSC lines, but not in BCFF cells. Despite the presence of FZD1, GNA12, RHOA, and TAZ, the critical downstream factors YAP and TEAD4 were not detected in BCFF cells. TEAD4, a downstream target of WNT-YAP/TAZ signaling, promotes the expression of CDX2 and plays an important role in the self-renewal and maintenance of TSCs in vitro (Nishioka et al., 2008, 2009; Ralston et al., 2010). Bovine TSCs established using Wnt5a, an alternative ligand of the WNT-YAP/TAZ pathway that is incapable of activating canonical WNT signaling, demonstrated typical TSC morphology (data not shown). However, these cells grew at a slower rate relative to the BTSW cells, suggesting that WNT-YAP/TAZ signaling is necessary for the in vitro establishment of bovine TSCs and that WNT/β-catenin signaling contributes to the maintenance of stemness.
Conclusions
Using more stable and controllable modified culturing systems utilizing the addition of Wnt3a, we have established bovine TSC lines and demonstrated that their properties are maintained by Wnt3a by regulation of CDX2 expression through the WNT-YAP/TAZ signaling pathway. One of the established cell lines exhibited characteristics that were indicative of an intermediate state between ESCs and TSCs. Our results also demonstrated that Wnt3a not only maintained bovine TSC pluripotency but also promoted the expression of specific TSC marker genes and maintained TSC characteristic.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31460309), the National Transgenic Project of China (2016ZX08010001-002 and 2016ZX08010005-001), and the Science and Technology Program of Inner Mongolia, China (201502073).
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Video S1
Supplementary Video S2
Supplementary Video S3
Supplementary Video S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.