
Abstract
To perform global transcriptome profiling using RNA-seq in the peripheral blood of intracerebral hemorrhage (ICH) patients. In 11 patients with ICH, peripheral blood was collected within 24 h of symptom onset or last known well, and a second blood draw occurred 72 h (±6) after the first. RNA-seq identified differentially expressed genes (DEGs) between the first and second samples. Biological pathway enrichment analysis was performed with Ingenuity
Introduction
Intracerebral hemorrhage (ICH) is a severe neurological disorder with 30-day mortality of ∼40% (van Asch et al., 2010), and those who survive often have significant morbidity (Broderick et al., 1992). ICH accounts for 10% of strokes but 50% of stroke mortality (Benjamin et al., 2017). There are no medical or surgical therapies that have shown definitive benefit in ICH. Inflammation after ICH contributes to neuronal injury and poorer outcomes (Wang and Dore, 2007; Chen et al., 2015). Post-ICH inflammation has been previously described as occurring in four major stages: (1) localized inflammation, (2) activation of microglia, astrocytes, and breakdown of the blood–brain barrier, (3) infiltration of immune cells from the peripheral blood, and (4) anti-inflammatory and reparative mechanisms (Askenase and Sansing, 2016).
There is substantial uncertainty regarding the implicated immune cells, the interactions between these cells, and the overall time course of the inflammatory response, including when and how a transition may occur from damaging to reparative mechanisms. White blood cells (WBCs) play an important role in post-ICH inflammation (Wang and Dore, 2007; Wang, 2010), and increasing evidence suggests that monocytes, a specific type of WBCs, are particularly important (Adeoye et al., 2014; Hammond et al., 2014a, 2014b; Walsh et al., 2015a, 2015b; Morotti et al., 2016). We previously reported in two independent patient cohorts that higher peripheral blood monocyte count was associated with greater ICH 30-day case fatality (Adeoye et al., 2014; Walsh et al., 2015a).
However, as stated in a recent review article that described stages of inflammation after ICH, “It is apparent how much we have yet to understand” (Askenase and Sansing, 2016). Next-generation sequencing-based RNA-seq is a powerful method that provides a global unbiased transcriptomic analysis. RNA-seq also has high sensitivity and specificity, and it can identify more of the overall differentially expressed genes (DEGs) compared with other methods for characterizing gene expression (Li et al., 2016). Findings from RNA-seq have shown increasing promise for the study of disease and clinical management (Deng et al., 2006; Coates et al., 2015; Van Allen et al., 2015; Byron et al., 2016).
In this report, we performed RNA-seq in peripheral blood samples from ICH patients from whom the first blood sample was collected within 24 h of symptom onset or last known well. We hypothesized that ICH would increase the expression of inflammation-related genes and induce novel biological pathways.
Materials and Methods
Patient enrollment
Cases were enrolled between November 2017 and January 2018 at an urban tertiary care hospital. Study procedures were approved by the University of Cincinnati Institutional Review Board (IRB Study ID 2016-7230). Potential cases were identified from pages to the Stroke Team and 24/7 screening of the emergency department, neurology/neurosurgery floor beds, and neuroscience intensive care unit by hospital-based research coordinators.
Inclusion criteria were as follows: age ≥18 years, history and radiographic findings consistent with primary spontaneous ICH, and enrollment within 24 h of last known normal or witnessed symptom onset. Exclusion criteria included ruptured aneurysm, arteriovenous malformation, or vascular anomaly, traumatic etiology of ICH, hemorrhagic conversion of an ischemic stroke, recurrence of a recent (<1 year) hemorrhage, known active cancer or brain metastases, current status of being incarcerated, in police custody, or institutionalized. The patient or the legally authorized representative provided signed informed consent and were then asked about medical history related to stroke risk factors. Venipuncture was performed for collection of a venous blood specimen.
Sample collection
The first peripheral blood sample was obtained from each patient within 24 h of ICH symptom onset or last known normal. A second peripheral blood sample was collected from each patient 72 h (±6 h) after the first. At the time of each blood draw, 2.5 mL of blood was collected in PAXgene® (Preanalytix, Hombrechtikon, Switzerland) blood RNA tubes. Consistent with the manufacturer's instructions, the tubes were gently inverted 8 to 10 times, stored at room temperature for 2 h, and then frozen. The samples were stored at −80°C until the time of RNA extraction.
RNA extraction and Globin-Zero RNA-seq
The experiment was performed by the genomics, epigenomics, and sequencing core at the University of Cincinnati. Total RNA was extracted using PAXgene Blood RNA Kit (Qiagen, Hilden, Germany), including DNase I treatment according to the manufacturer's instructions. The eluted RNA was again treated with DNase I and cleaned using the RNA Clean & Concentrator kit (Zymo, Irvine, CA) to remove trace amounts of genomic DNA. The RNA quality was measured by using Bioanalyzer RNA 6000 Nano Kit (Agilent, Santa Clara, CA). Globin-Zero Gold kit (Illumina, San Diego, CA) was used to enrich nonglobin coding and noncoding RNAs from blood-derived total RNA.
To ensure high reproducibility of the process, the automated SMARTer Apollo system (Takara, Mountain View, CA) was used to deplete globin and rRNA from 1 μg total RNA. To prepare the library for RNA-seq, NEBNext Ultra Directional RNA Library Prep kit (New England BioLabs, Ipswich, MA) was used (Sharma et al., 2017). After indexing and enrichment through 11 cycles of PCR, the libraries together with the negative control were cleaned by AMPure XP beads for Bioanalyzer QC analysis.
To measure the library concentration, NEBNext Library Quant Kit (New England BioLabs) together with QuantStudio 5 Real-Time PCR Systems (Thermo Fisher, Waltham, MA) was used. Pooled libraries at the final concentration of 15 pM was clustered onto an single-read (SR) flow cell using Illumina TruSeq SR Cluster kit v3. RNA-seq was performed using the Illumina HiSeq 1000 with the sequencing setting of single read 1 × 51 bp to generate ∼50 M reads per sample. The RNA-seq data from the study are available through the Gene Expression Omnibus (GEO) of the National Center for Biotechnology Information (NCBI), record number GSE125512.
Determination of DEGs and enriched biological pathways
To identify differential gene expression, sequence reads were aligned to the reference genome GRCh37 by using standard Illumina sequence analysis pipeline and then analyzed by The Laboratory for Statistical Genomics and Systems Biology at the University of Cincinnati. The aligned reads were quantified and converted to relative gene expression level represented by FPKM (fragments per kilobase million). The differential expression was analyzed by comparing the second sample with the first using the DESeq2 Bioconductor package. Specifically, the RNA-seq read counts were modeled following a negative binomial distribution and fit by a generalized linear model for each gene. Wald test was applied on the fitting coefficients to test for differential expression. The Benjamini–Hochberg procedure was applied on the p-values for multiple testing to estimate false discovery rate (FDR).
The significance of DEGs was defined by adjusted p-value (FDR) less than 0.1. FDR <0.1 is a conventional threshold for significance when analyzing RNA-seq data as reported in a publication regarding DESeq2 methodology (Love et al., 2014) as well as a number of transcriptional investigations (Cabezas-Wallscheid et al., 2014; Bergsveinson et al., 2016; Nagaraja et al., 2017; Pantazatos et al., 2017).
To interpret the gene expression data, Ingenuity
IPA compares the experimental data set with many defined canonical pathways, disease/function annotations, and upstream regulators, each serving as a hypothesis for the biological mechanisms for the data being analyzed. Each of these options is then assigned two statistical scores: a p-value and a z-score.
More specifically, to measure the likelihood that an association between DEGs from the data set and a given IPA category is due to random chance, IPA utilizes a right-tailed Fisher's exact test with a p-value <0.05 defining a statistically significant nonrandom association with over-representation of focus genes in that pathway, disease/function, or regulator. In addition to statistical significance, the IPA z-score also predicts the direction of change for a given pathway, disease/function, or regulator that is calculated based on the data set's correlation with the activated state of that given IPA biological variable. A z-score greater than 2 indicates significantly upregulated functional activity, and a z-score less than −2 indicates significant downregulation.
Results
Eleven ICH patients were enrolled and demographic information and past medical history were recorded (Table 1, Supplementary Table S1). A total of 16,640 genes were identified by RNA-seq. There were 218 genes that were differentially expressed between the second and first samples (FDR <0.1, Supplementary Table S2). All samples showed high-quality RNA (RNA Integrity Number ∼9).
Demographic Information and Medical History for Intracerebral Hemorrhage Patients (n = 11)
Time to first blood draw for the study relative to time of witnessed ICH symptom onset or last known well.
COPD, chronic obstructive pulmonary disease; ICH, intracerebral hemorrhage; IQR, interquartile range; SD, standard deviation.
IPA identified 97 annotated categories of diseases and functions that were significantly upregulated (z-score >2.0) and included a total of 188 of the DEGs from the study. Annotated diseases and functions specifically related to activation of immune system cells accounted for 46 of the 97 categories (e.g., chemotaxis of leukocytes, binding of mononuclear leukocytes, adhesion of granulocytes, and migration of mononuclear leukocytes). Of these 46 annotations for immune cell activation, there were 13 specifically related to activity of phagocytic cells (e.g., cell movement of macrophages, binding of macrophages, and response of phagocytes).
Out of the 97 total upregulated categories, 22 compromised annotations for more general cellular activation processes (e.g., cell movement, migration of cells, homing of cells, chemotaxis, invasion of cells, microtubule dynamics, cell cycle progression, and formation of cellular protrusions). Finally, there were four annotations for other inflammation-related processes (e.g., inflammatory response). In contrast to the 97 annotated categories that were significantly upregulated, only 9 were predicted to be significantly downregulated (z-score less than −2.0).
Within the top two networks of molecular interactions that were predicted by IPA (scores of 37 and 32, respectively), there were substantial nodes with a large number of connections (edges) with other molecules. In the first network, extracellular signal regulated kinases 1 and 2 (ERK1/2) were the most highly connected node, followed by integrin beta 3 (ITGB3) (Fig. 1). For the second network, NF-κB was the most highly connected node (Fig. 2).

The top network of molecular interactions predicted by IPA based on the reported RNA-seq data (consistency score 37). The node with the most connections with other molecules is ERK1/2. The node with the second most connections with other molecules is ITGB3. ERK1/2, extracellular signal regulated kinases 1 and 2; IPA, ingenuity pathway analysis; ITGB3, integrin beta 3.
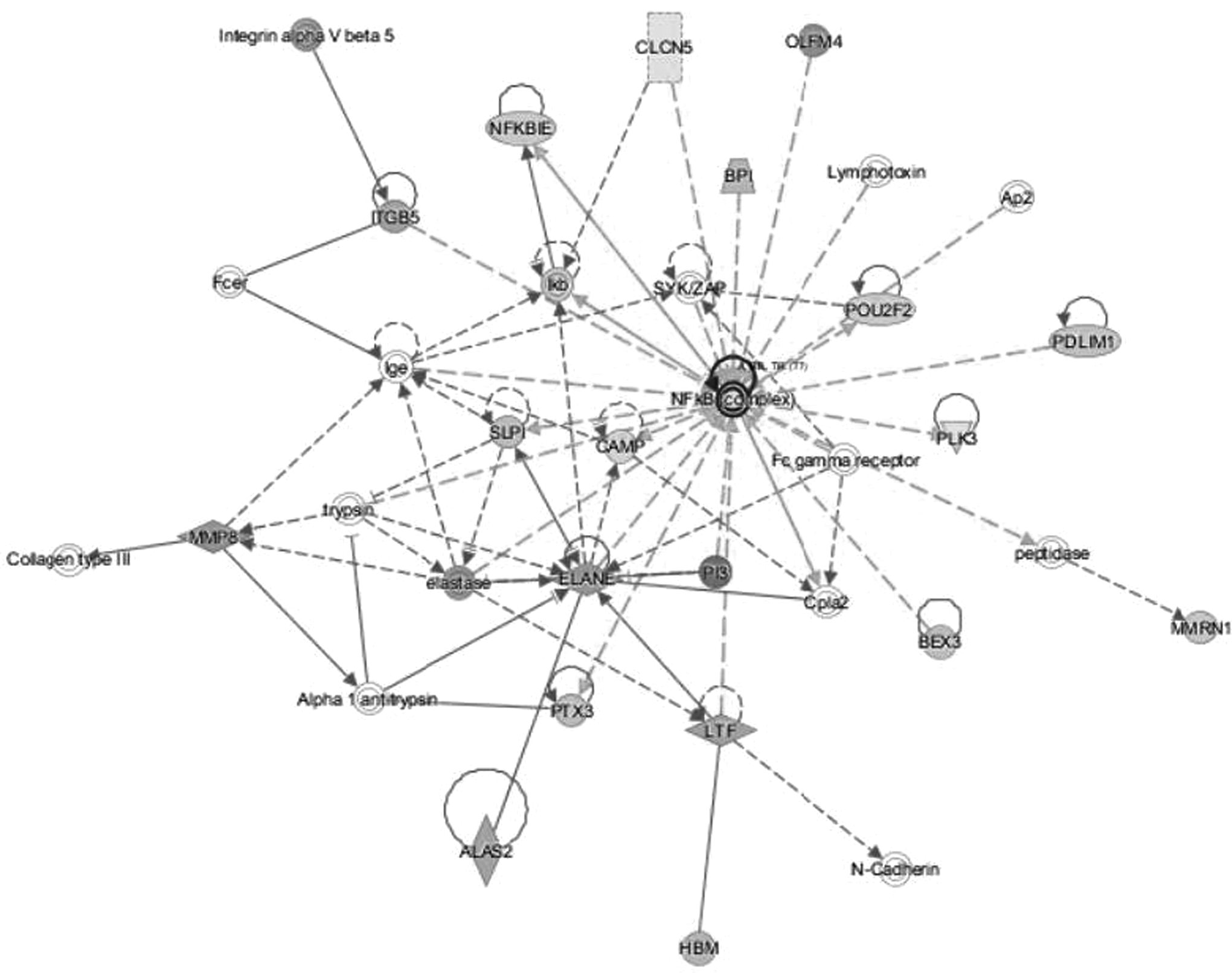
The second significant network of molecular interactions predicted by IPA based on the reported RNA-seq data (consistency score 32). The node with the most connections with other molecules is NF-κB.
IPA identified canonical pathways, and the most statistically significantly upregulated pathways included interleukin (IL)-8 signaling (z-score 3.00, p-value 8.44E-5) and NF-κB activation (2.55, 1.78E-4). Within these pathways, there were 12 unique DEGs from our RNA-seq data, all of which were upregulated in the second ICH sample compared with the first (Table 2).
Differentially Expressed Genes That Comprise Two of the Most Significantly Upregulated Canonical Pathways from Ingenuity Pathway Analysis
IL-8, interleukin-8 signaling; NF-κB, nuclear factor kappa B activation.
For upstream regulator analysis, six were identified as having the lowest p-values and to also have statistically significantly activated or inhibited states (defined as z-score of less than −2.0 as inhibited and >2.0 as activated). The upstream regulators were defined by IPA as a chemical drug, biologic drug, cytokine, growth factor, or transcription regulator. The two upstream regulators that were predicted to be inhibited were the chemical drug dexamethasone (z-score −2.69, p = 5.93E-21) and the biologic drug filgrastim (−4.68, 6.78E-17). The four upstream regulators that were predicted to be activated were the chemical drug lipopolysaccharide (LPS) (3.38, 3.17E-16), the cytokine TNF (2.37, 6.65E-9), the transforming growth factor beta 1 (TGFβ1) (3.31, 2.17E-8), and the transcription regulator CCAAT enhancer binding protein epsilon (CEBPE) (2.22, 2.47E-8).
Discussion
The current study is, to the best of our knowledge, the largest reported to date of RNA-seq in ICH patients. We found that there are significant changes in peripheral blood gene expression at 72 to 96 hours compared with 0 to 24 hours from ICH symptom onset. Numerous components of the bioinformatics analysis through IPA supported the concept that these induced genes were primarily related to activation of the immune system and other inflammatory and cellular proliferation processes. Mediators of particular importance, discussed further hereunder, included IL-8, NF-κB, ERK1/2, integrins, and predicted upstream regulators.
One of the canonical pathways determined by IPA to be most significantly upregulated was IL-8 signaling. IL-8 is a proinflammatory cytokine that was increased in severe sepsis compared with uncomplicated sepsis both in the emergency department (p = 0.0009) and up to 24 h after presentation (p = 0.011) (Macdonald et al., 2014). When IL-8 was studied in 76 ischemic stroke patients compared with 28 controls, it was increased in stroke patients' plasma within 24 h of symptom onset (p < 0.001), and IL-8 levels were also positively associated with the extent of the ischemic stroke lesion (p < 0.01) (Domac and Misirli, 2008).
The inhibition of IL-8 as a therapeutic target for stroke has been studied, including its inhibition by antiplatelet agents in ischemic stroke patients (Al-Bahrani et al., 2007), reduced inflammation and neurological deficits after treatment with the IL-8 inhibitor reparixin in rats with transient cerebral ischemia (Villa et al., 2007), and less edema and infarct size in a rabbit model of transient brain ischemia after treatment with an IL-8 neutralizing antibody (Matsumoto et al., 1997). Although data regarding IL-8 and ICH are limited, in 94 patients with basal ganglia ICH, a positive correlation was reported between IL-8 and severity of cerebral edema (r = 0.305, p < 0.05) (Wang et al., 2016).
A canonical pathway associated with NF-κB activation was also significantly upregulated. NF-κB, a transcription factor that has been reported as a critical mediator of neuroinflammation-associated pathophysiology (Shih et al., 2015), was also the most highly connected node in an IPA-generated network based on the reported DEGs (Fig. 2). NF-κB has an important role in a number of cellular functions including inflammation, immune responses, and cellular proliferation (Kaltschmidt et al., 2005; Ledoux and Perkins, 2014). It is activated by many factors such as inflammatory stimuli, cytokines, bacterial toxins, viruses, oxidative stress, and by specific molecules such as LPS (Baldwin, 1996). NF-κB then regulates the expression of almost 500 different genes including the cytokines IL-8 and TNF, noted in the current study, enzymes, cell cycle regulators, and promoters of angiogenesis (Gupta et al., 2010).
NF-κB has been studied in the context of immune responses and ischemic stroke (Harari and Liao, 2010). In a rat model of ischemic stroke, neuregulin-1 produced neuroprotective and anti-inflammatory effects through differential regulation of NF-κB signaling pathways in microglia (Simmons et al., 2016). The peroxisome proliferator-activated receptor gamma (PPAR-γ) transcription factor, considered for ICH treatment due to anti-inflammatory effects, likely mediates some of these effects through NF-κB (Zhao et al., 2015). In a murine model of ICH, NF-κB regulation has been implicated in activation of autophagy and associated neuronal damage (Shen et al., 2016). In 53 ICH patients, the perihematomal region was analyzed with immunohistochemistry and found to have higher levels of NF-κB (p < 0.001) relative to controls. The sample collection time varied from <6 to >96 h from symptom onset, and NF-κB levels peaked from 13 to 24 h (Zhang et al., 2014).
The top network of molecular interactions that was generated by IPA included ERK 1/2 as having the most interactions with other molecules (Fig. 1). ERK 1/2 are protein serine/threonine kinases, members of the MAPK family that participate in a variety of processes such as cell differentiation, metabolism, proliferation, adhesion, and inflammatory processes (Roskoski, 2012). In human monocytic cells (THP-1), ERK signaling mediated the production of inflammatory cytokines such as IL-1β and TNF-α (Kurosawa et al., 2000). In intestinal epithelial cells, bacteroides fragilis enterotoxin increased production of IL-8 versus controls (p < 0.01) through a mechanism involving simultaneous activation of ERK and NF-κB (Wu et al., 2004).
In epithelial cells infected with chlamydia trachomatis, IL-8 production was dependent on ERK signaling (Buchholz and Stephens, 2007). The ERK 1/2 pathway has been reported as a potential therapeutic target for neurological diseases, including stroke (Sun and Nan, 2017). In rats with ischemic stroke, production of inflammatory cytokines such as TNF-α and IL-6 was transcriptionally regulated through ERK, and injection of an ERK1/2 inhibitor at 0 and 6 h after stroke reduced infarct volume (11.7% and 15% of total brain volume, respectively, compared with 25% for controls) (Maddahi and Edvinsson, 2010). Similar results have been reported from preclinical models of subarachnoid hemorrhage, including that inhibition of ERK signaling resulted in less expression of proinflammatory cytokines such as IL-6 (p < 0.01) and IL-1 beta (p < 0.05) (Maddahi et al., 2011).
In ICH, preclinical studies have been reported such as that hemolysate in astrocytes increased the phosphorylation of ERK 15-fold compared with controls (p < 0.01) (Yang et al., 2016). In a rat model, ICH increased ERK expression in perihematomal tissues (Wen et al., 2017).
Members of the integrin class of cell surface receptors were DEGs in significantly upregulated canonical pathways (Table 2) and major components of a network (Fig. 1). The three integrins that were DEGs in our study were among those genes with the largest fold changes in the canonical pathways and included the following: integrin β3 (Fig. 1 and Table 2), integrin α2b (Fig. 1 and Table 2), and integrin β5 (Table 2). Within the IPA-generated network (Fig. 1), there were a total of four integrins: β3, α2b, α3β1, and α4β1. The integrins consist of noncovalently associated α and β subunits (Campbell and Humphries, 2011).
Integrin signaling is associated with various pathophysiological processes such as inflammation, autoimmunity, and malignancy (Anderson et al., 2014). Integrins have been studied on leukocytes and noted to change over time and in response to various signals. For example, human monocytes expressed β1 and β2, but αVβ3 was induced through differentiation into macrophages (Harris et al., 2000). The αVβ3 integrin regulated the inflammatory responses of macrophages in an in vitro model, specifically, activation of αVβ3 caused increased expression of inflammatory cytokine mRNA including TNF-α (8- to 28-fold), IL-1β (15- to 30-fold), IL-6 (2- to 4-fold), IL-8 (5- to 15-fold), and significant suppression of the anti-inflammatory cytokine IL-10 (Antonov et al., 2011).
In a murine model of ICH, α4 integrin was elevated on all leukocyte populations (p < 0.05), αLβ2 was only increased on T cells (p < 0.05), and an α4 antagonist resulted in less leukocyte infiltration into the brain and less neurobehavioral disability (p < 0.01) (Hammond et al., 2014a).
Upstream regulators are identified by IPA with the goal that these factors explain the “downstream” gene expression. Downstream gene expression in this context refers to those DEGs reported as part of a given experimental data set. Thus, IPA utilized information in the Ingenuity Knowledge Base, that is, the reported literature regarding the expected effects between transcriptional regulators and their target genes, to identify upstream regulators that would explain the gene expression in our ICH study.
The upstream regulators predicted from our findings supported the concept that ICH induced inflammatory pathways in peripheral blood, more specifically, anti-inflammatory regulators were inhibited, and inflammatory regulators were activated. Dexamethasone, identified as a significantly inhibited upstream regulator, is a corticosteroid with numerous anti-inflammatory effects (Kaneguchi et al., 2018).
The medication filgrastim, the second significantly inhibited upstream regulator, is a granulocyte colony-stimulating factor that is better known for stimulating the production of leukocytes. However, filgrastim also has a number of anti-inflammatory properties such as reducing the production of the inflammatory cytokines TNF-α, IL-12, and IFN-γ (Hartung et al., 1999), and increased production of anti-inflammatory counterregulators such as IL-1 receptor antagonist and soluble TNF receptors (Hartung et al., 1995).
The activated upstream regulators in our study, LPS, TNF, CCAAT, and TGFβ1, are associated with promoting inflammatory and immune system-related downstream events. LPS, also known as endotoxin, is a major component of the outer membrane of gram-negative bacteria and induces a cascade of cellular reactions that potentiates a strong inflammatory response. TNF is a well-known cytokine that induces systemic inflammation and participates in the acute phase response, a complex early defense system that can be activated by a number of events such as infection, inflammation, and trauma (Bradley, 2008). CCAAT is a transcription factor that has also been implicated in promoting inflammation, including inflammatory responses in macrophages (Rahman et al., 2012).
TGFβ1 was first reported to be a potent cytokine that initiates inflammation. Although subsequent findings have supported both inflammatory and anti-inflammatory effects (Han et al., 2012), TGFβ1 is known to be a powerful chemotactic agent, and it stimulates migration of cells such as monocytes, neutrophils, and lymphocytes with only 10−15 M concentrations (McCartney-Francis and Wahl, 1994). Increased TGFβ1 has also been implicated in microglia-mediated inflammation after ICH and subsequent functional recovery (Taylor et al., 2017).
In previous studies from our core sequencing facility, an alternative approach of quantitative reverse transcription polymerase chain reaction (RT-qPCR) was used to validate DEGs (Sharma et al., 2017). This showed that very high reproducibility between RNA-seq and RT-qPCR results. In this study, the same methods and reagents were used under the same experimental conditions, thus, no additional validation of the RNA-seq result was performed.
Overall, the existing literature regarding post-ICH neuroinflammation includes a predominance of preclinical studies (Moxon-Emre and Schlichter, 2011; Sansing et al., 2011; Hammond et al., 2014a, 2014b; Mracsko et al., 2014; Zhang et al., 2017). Cells that have been implicated in the peripheral blood inflammatory response after ICH include monocytes, T cells, and neutrophils.
In a murine ICH study, monocytes secreted TNF, and mice with fewer of these monocytes had better motor function than controls (Hammond et al., 2014b). In other rodent investigations, CD4+ T cells were the predominant leukocyte to infiltrate from 1 to 14 days from ICH induction (Mracsko et al., 2014) and damaged the blood brain barrier (Zhang et al., 2017). ICH-associated cerebral inflammation was attenuated, at least in part, through reduction of T cell infiltration (Rolland et al., 2013; Liu et al., 2016).
Neutrophils become highly activated in the perihematomal region. Inflammatory cytokines and other factors bind to neutrophils resulting in the production of inflammatory molecules such as cytokines, reactive oxygen species, and nitric oxide, all of which contribute to tissue damage (Keep et al., 2012). A number of studies that have inhibited or depleted neutrophils or monocytes have consistently decreased inflammation and improved neurological functional outcomes, providing further evidence for a harmful role of these cells in the early period after ICH (Moxon-Emre and Schlichter, 2011; Sansing et al., 2011).
Clinical studies of post-ICH inflammation that do not study gene expression tend to report cell counts or a limited number of other predetermined cellular biomarkers (Adeoye et al., 2014; Hammond et al., 2014b; Walsh et al., 2015a; Morotti et al., 2016; Su et al., 2017). We note that our findings are consistent with these previous reports that ICH is associated with or induces inflammatory changes in peripheral blood. This includes that greater ICH mortality (Adeoye et al., 2014; Walsh et al., 2015a) and hematoma expansion (Morotti et al., 2016) were associated with higher peripheral blood monocyte count. Higher peripheral blood levels of CCL2, a major chemokine for monocyte recruitment, were associated with poor functional outcome in ICH patients (Hammond et al., 2014b). In ICH patients, a higher CD4+/CD8+ T cell ratio was associated with elevated intracranial pressure and worse Glasgow Outcome Scale at 30 days (Su et al., 2017).
To the best of our knowledge, there are three published reports of RNA-seq in human ICH. Sang et al. reported 4040 DEGs in peripheral blood mononuclear cells from four ICH patients compared with four controls. Blood was drawn within 3 h of symptom onset, providing evidence that ICH induces changes in peripheral blood very soon after the hemorrhage (Sang et al., 2017).
Dykstra-Aiello et al. analyzed whole blood with RNA-seq in 4 patients with ICH, 12 with ischemic stroke, and 4 controls, and found differential alternative splicing for 412 genes among the groups (FDR p < 0.05), including those involved in cell death, cell survival, and cellular immune response. Distinct expression signatures from 292 genes differentiated the stroke groups (p < 0.0005) (Dykstra-Aiello et al., 2015). Most recently, Goods et al. investigated the handling and storage of blood and leukocyte isolation methods and how these affected the global transcriptome of immune cells. For one of the many aims of this publication, blood was analyzed with RNA-seq from three ICH patients and six controls (Goods et al., 2018).
Our investigation is novel in a number of aspects compared with the three previously published studies of RNA-seq in human ICH. With 11 ICH patients, this report has the largest sample size compared with 4 (Dykstra-Aiello et al., 2015), 4 (Sang et al., 2017), and 3 (Goods et al., 2018) ICH patients. Ours is the first study to analyze serial samples from the same ICH patients. Instead, a single sample from ICH patients was compared with controls in the other three investigations (Dykstra-Aiello et al., 2015; Sang et al., 2017; Goods et al., 2018). By studying transcriptomic changes in the same patients over time, we minimized individual variation that can exist between ICH cases and controls, despite matching, and we strengthened the potential to report transcriptomic changes secondary to post-ICH pathophysiology.
Our study included a specific time range for initial enrollment and sample collection that was in the acute phase from ICH onset (within 24 h). This differed from other reports, such as a large range of 5.8 to 101.2 h from stroke onset to enrollment (Dykstra-Aiello et al., 2015). Our investigation analyzed ICH patients only, compared with, for example, a report in which there were three times the number of ischemic stroke patients compared with ICH (Dykstra-Aiello et al., 2015). We enrolled both African American and Caucasian ICH patients, as opposed to prior investigations that enrolled only Caucasian males (Dykstra-Aiello et al., 2015) or did not report the race/ethnicity of the ICH subjects (Sang et al., 2017; Goods et al., 2018).
We analyzed the peripheral blood of ICH patients with the objective of identifying DEGs and enriched biological canonical pathways, disease/function annotations, upstream regulators, and molecular networks using IPA. This methodology is unique compared with the existing publications for which the focus was instead differential alternative splicing (Dykstra-Aiello et al., 2015), transcriptional differences due to sample processing and transport (Goods et al., 2018), and enrichment analysis with other bioinformatics tools such as DAVID and KEGG (Sang et al., 2017).
Finally, compared with other studies of RNA-seq in human ICH, in which more general categories of cellular and immune system activation were reported, our study is the first to report molecular networks and to implicate specific mediators of post-ICH neuroinflammation, including IL-8, NF-κB, ERK 1/2, and members of the integrin class as discussed throughout this article.
In addition to novel components of our study as already discussed, we note additional strengths. Our results demonstrated that ICH induces systemic changes in inflammatory gene expression that have potential as novel pathophysiologic biomarkers and treatment targets. By investigating human ICH, our study avoids the limitations of preclinical ICH models and reports findings from ICH patients that can both advance this line of inquiry generally and inform reverse translation for the research of molecular mediators of post-ICH neuroinflammation in preclinical models. Through utilizing RNA-seq, we report findings from unbiased global transcriptional profiling with the potential to identify unanticipated molecular mediators and enriched biological pathways. By performing RNA-seq in peripheral blood, obtained regularly in the clinical setting, our findings have the potential for more translational relevance. Inflammatory pathophysiologic biomarkers are also more likely to be modifiable and amenable to treatment.
Limitations of our reported research include a relatively small sample size, under-representation of females, lack of control subjects, and the exploratory nature of the study. We were not able to assess for associations between RNA-seq findings and ICH severity or clinical outcome. Clinical information regarding the patient's recovery status was not available.
RNA-seq data were not analyzed more than ∼96 h from symptom onset. Although the focus of this study was on the acute period after ICH, analysis of samples collected later, such as a week from stroke onset, could allow for characterization of unique gene expression over a longer period of time during which anti-inflammatory and reparative mechanisms might begin to predominate. Furthermore, although all patients were enrolled within 24 h of ICH onset or last known well, investigating the gene expression at earlier time points could also strengthen the study. For example, the enrollment of all patients within 6 or 12 h of symptom onset could provide more insight into the hyperacute period after ICH.
Conclusion
We found that gene expression in peripheral blood is induced at 72 to 96 h compared with 0 to 24 h from ICH symptom onset, and the associated biological pathways are primarily those associated with inflammation and immune system activation. Inflammatory mediators of particular relevance that were upregulated by ICH included IL-8, NF-κB, ERK 1/2, and members of the integrin class.
Our findings build on our prior work regarding peripheral blood leukocytes and ICH and contribute to the larger body of literature regarding neuroinflammation after ICH. Further research is needed, including larger sample sizes, additional serial blood samples drawn earlier after symptom onset and extending for a longer period of time, analysis of matched control subjects, and association of transcriptomic findings with markers of ICH severity and clinical outcome. Overall, additional research is warranted with the goal of better understanding post-ICH inflammatory pathophysiology and identifying therapeutic targets for this devastating condition with no current treatment.
Footnotes
Acknowledgment
The authors acknowledge the financial support for the reported research from the Mayfield Education and Research Foundation/University of Cincinnati Department of Neurosurgery Research Grant Program, Cincinnati, Ohio.
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.