
Abstract
The present study aims to identify the species and strains of Mycoplasma hyopneumoniae isolated from Tibetan pigs (Mh TB1) at the genetic level for understanding the basis of its pathogenicity. Mh TB1 was isolated from the consolidated lungs of Tibetan pigs by liquid culture and agar plate colony method. Polymerase chain reaction (PCR) amplification of the 16S recombinant DNA (rDNA) conservative sequence and a species-specific gene (P36) of Mh provided species confirmation. PCR products were imaged on gels and shotgun sequencing was performed. DNA sequences were compared for assessing genetic similarity between Mh TB1 and Mh reference strains in the GenBank database. The isolated strains were >98% similar to the Mh reference strains. Genomic analysis revealed significant sequence conservation between Mh TB1 and the reference strains; however, differential genes were more prevalent in Mh TB1 than in other reported strains. Therefore, we concluded that Mh is a major pathogen of Tibetan pigs that cause enzootic pneumonia. The Mh TB1 strain harbors more genes and specific virulence factors, consistent with its plateau-related adaptability to hypoxia and virulence. Differential gene analysis revealed gene variations in the inclement plateau environment, enriched gene pool, and plateau adaptability of the Mh TB1 strain, which will be important for vaccine development.
Introduction
Tibetan Pig is a native breed of pig in Qinghai-Tibet Plateau. The Tibetan Pigs are mainly distributed in the eastern part of Tibet, Yunnan, Qinghai, and Sichuan Province, People's Republic of China. The major habitat of the Tibetan Pig breed is the forest and the valley zone that is 2,900–4,100 m above sea level.
Tibetan pigs are raised by grazing. The total number of Tibetan pigs is about 300,000. There are ∼4,000 purebred Tibetan pigs in the four provinces. Now, the distribution regions of the purebred Tibetan pig are decreasing and the Tibetan pig breed is experiencing the process of extinction. According to Article 12 of the Animal Husbandry Law, the Ministry of Agriculture identified Tibetan pig (local breed 48) as the national animal and poultry genetic resource protection variety.
The Tibetan pig breeds have a relatively slow growth rate when compared with Landrace. Adult Tibetan pigs (about 2 years) generally weigh not more than 50 kg. The value of biological genetic resources and economic value of Tibetan pigs is relatively large. With the increase of the number of factory farming pigs, the importance of ecological health culture and disease prevention and control of this breed is increasingly prominent. Due to the particular geographical and climatic conditions of the plateau, and the lack of attention to the Tibetan pigs in the traditional way of life of the herdsmen, the increase in the number of foreign pigs and the rise of factory farming have affected the Tibetan pig herd. The Tibetan pig mycoplasma pneumonia prevention and control has been in a blank state for a long time and needs to be paid enough attention.
In summary, the Tibetan pig is a plateau miniature pig breed, an important source of animal husbandry with genetic and economic value. Porcine respiratory disease is an expensive health concern in swine-producing areas worldwide (Meens et al., 2006; Charlebois et al., 2014). Mycoplasma hyopneumoniae (Mh) is a major contributor and predisposing factor in these cases (Armstrong et al., 1987; Madsen et al., 2007). Mh exposure results in the establishment of secondary bacterial infections that are important in the porcine respiratory disease complex and enzootic pneumonia and have been associated with the potentiation of other pathogenic conditions such as porcine reproductive and respiratory syndrome and pneumonia caused by porcine circovirus type 2. Mh is the primary agent of swine enzootic pneumonia, which has high morbidity and low mortality rates (Stemke, 1997), asthma, and slow growth (L'Ecuyer and Boulanger, 1970). Epidemiological data indicate that the Mh antibody-positive rate in Tibetan pig herds in 2014 was 73% (Qiu et al., 2018), as shown by enzyme-linked immunosorbent assay. Polymerase chain reaction (PCR) and gel electrophoresis of bronchoalveolar lavage fluid revealed that 6% of the slaughtered Tibetan pigs were Mh positive. This indicates that a certain proportion of Tibetan pigs had been infected and had recovered, or were recessive bacteria carriers (Marois et al., 2010; Takeuti et al., 2017).
Infections in herds should be monitored. Isolation and identification of Mh is still the golden standard in disease diagnosis (Maurício et al., 2015). However, isolation of this pathogen is a long cycle and easy to be disturbed by the vigorous growth of Mycoplasma hyorhinis. Advances in Mh research have been hampered by the organism's fastidious growth conditions and the lack of genetic tools and transformation protocols (Wei et al., 2013). Therefore, only few virulence determinants or virulence-associated determinants have been identified. Bacterial contamination and concealment of M. hyorhinis necessitates Mh separation in more than three repeats of sterile medium filtration and addition of specific antiserum to the medium to yield culture-positive specimens. Sequencing of the 16S recombinant DNA (rDNA) as the most commonly used molecular clock (Artiushin et al., 1993; Stemke et al., 1994; Stärk et al., 1998; Strait et al., 2008; Yamaguti et al., 2015) is the basis of accurate bacterial classification. This method reveals differences between species (Minion et al., 2004) and the sequencing technology is relatively easy to operate.
The aim of this study was to isolate Mh from the lungs of Tibetan pigs, and positively identify the bacteria using 16S rDNA and species-specific gene P36 (L'Ecuyer and Boulanger, 1970; Kwon and Chae, 1999; Liu et al., 2015) in PCR (Bej et al., 1991; Soini et al., 1992; Artiushin and Minion, 1996; Blanchard et al., 1996; Sørensen et al., 1997; Baumeister et al., 1998; Caron et al., 2000; Cai et al., 2007), agarose gel electrophoresis (AGE), sequencing, morphology analysis, and DNA sequence analyses. We also performed whole-genome sequencing of Mh TB1 and compared its sequence with other Mh reference sequences to understand the genetic basis of its pathogenicity.
Materials and Methods
Ethics statement
The animal experiments and procedures were performed in strict accordance with the pertinent guidelines (Proclamation No. 5 of the Standing Committee of Hubei People's Congress, P.R. China). The protocol was approved by the Committee on the Ethics of Animal Experiments at the College of Veterinary Medicine, Huazhong Agricultural University (approval permit No. 4200695758).
Sample collection, isolation, and identification of M. hyopneumoniae from Tibetan pigs
Sample collection
The age of the Tibetan pigs was between 6 and 18 months. There were 53 male and 67 female Tibetan pigs. Slaughter involved stunning by the application of 190 V of electricity, followed by bleeding upon severing the major blood vessels of the heart. Lung tissue samples were obtained rapidly from the pigs after the animals had been sacrificed. Samples were collected from the lungs of 120 Tibetan pigs in 2 abattoirs in Nyingchi, Tibet, P.R. China. The main clinical signs of Tibetan pig enzootic pneumonia were cough, asthma, labored breathing, slow growth, or one or more of these symptoms. Materials were obtained from pigs that showed typical pathological changes. The lesions were mainly distributed in the lung, lung lymph nodes, and mediastinal lymph nodes in both lungs, and had a consolidated appearance like fresh muscle. Samples were collected from the border with healthy lung tissue during dissection of the carcasses, vacuum packed in polypropylene vacuum bags, and stored at 4°C until 2 h after slaughter, after which Mh isolation was performed.
Isolation
One gram of lung sample from each pig was aseptically collected, washed with Hank's fluid to remove blood, and cut into ∼1 mm3-sized pieces. These were placed into KM2 basal culture medium, which is a modified FRIIS cell-free liquid medium (Topbiotech Co. Ltd., Zhaoyuan, P.R. China) with a pH of 7.6–7.8 (Hwang et al., 2010; Cook et al., 2016) containing 20% nonmycoplasma pig serum (treated at 56°C for 30 min; Gibco®), 50 μg/mL flucloxacillin (QiBo Chemical Co. Ltd, Shijiazhuang, P.R. China), 50 μg/mL cycloserine (Harveybio Co. Ltd., Beijing, P.R. China), 0.25% acetic acid thallium, 1% M. hyorhinis antiserum (homemade products), and 0.002% phenol red (Zhanyun Chemical Co. Ltd., Shanghai, P.R. China). Before the start of bacterial culture, the KM2 medium was filtered through a 0.22-μm filtration membrane and CO2 (5% in air) was introduced. The culture was incubated at 37°C. When the medium turned yellow and clear, one-fifth of the culture volume was removed, filtered through a 0.22-μm pore size membrane (Pilot Laboratory Equipment Co. Ltd., Tianjin, P.R. China) to remove contaminating bacteria, and fresh medium was added for the next passage. The remaining culture was preserved at −20°C after mixing 1:1 in 50% glycerol.
Identification
After five passages, PCR was used to amplify the 16S rDNA and the Mh species-specific gene P36 in the culture. Mh strain 7448 (GenBank: AE017244.1) was used as a reference strain. Table 1 presents the primers that were used. The primers were designed using National Center for Biotechnology Information (NCBI) Primer-basic local alignment search tool (BLAST) and synthesized by Sangon Biotech Co., Ltd. (Shanghai, P.R. China). Solid medium was made by adding 1% agar powder (Fuchen Chemical Co. Ltd., Tianjin, P.R. China) to the basal liquid medium. Single colonies were picked after growth on agar medium, diluted using physiological saline, and smears were made. Each smear was stained using Wright's and Giemsa stain and examined by optical microscopy.
Specific Primers of the 16S rDNA and P36 Genes Used for PCR
Bacterial liquid (2 mL) was centrifuged at 12,000 g for 3 min and the pellet was used for DNA extraction. Total genomic DNA was extracted using the DNA Extraction Reagent Kit (Tiangen Biotech Co., Ltd., Beijing, P.R. China) as detailed by the manufacturer. The extracted DNA was stored at −20°C until further processing. The Veriti® FAST thermal cycler PCR (Applied Biosystems, Foster City, CA) contained 12 μL of the PCR master mix (Beyotime Biotechnology, Shanghai, P.R. China), 0.5 μL (10 μmol/L) forward and reverse primers (Table 1), and 2 μL (23.5 μg/mL) DNA template. Double-distilled water was added to a final volume of 25 μL. For amplification of the 16S rDNA, the thermal cycling involved initial denaturation at 95°C for 5 min, 34 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 20 s, and a final extension at 72°C for 7 min. For amplification of P36, the thermal cycling involved initial denaturation at 95°C for 5 min, 34 cycles of 95°C for 30 s, 42°C for 30 s, and 72°C for 20 s, and a final extension at 72°C for 8 min. The PCR products were sequenced by the shotgun method (Quintara Biosciences, Wuhan, P.R. China).
AGE used Tris/acetic acid/EDTA (TAE) as the DNA electrophoresis buffer and 1% agarose (Biowest, Granada, Spain) containing 5 μL (per 100 mL) GoldView (Dingguo Changsheng Biotech Co. Ltd., Beijing, P.R. China). Electrophoresis was conducted at 120 V and 60 mA for 60 min.
The NCBI GenBank database and BLAST were used to construct phylogenetic trees using the MEGA6.0 software. The NCBI sequences of five Mh reference strains (M. hyopneumoniae 7448, 232, 168, J, and 7422) were used for neighbor-joining BLAST analysis to construct the phylogenetic trees.
Genomics analysis of M. hyopneumoniae from Tibetan pigs
One (TB1 strain) of eight isolated M. hyopneumoniae pathogenic strains from Tibetan pigs were used for comparative study. Genomic DNA was extracted and randomly fragmented. DNA fragments of the required length were obtained by electrophoresis. Adapters were ligated to the DNA fragments, followed by cluster preparation and sequencing.
Genome sequencing of pure cultures of Mh from Tibetan pigs was performed with a HiSeq 4000 sequencing platform (Illumina, San Diego, CA) at the Beijing Institute of Genomics using a next-generation sequencing system for the reversible terminator sequencing by the chemical synthesis method. The short reads were assembled into the genome sequence using SOAPdenovo 2.04. The assembly result was locally assembled and optimized according to paired-end reads and overlap relationship by mapping reads to Contigs. Gaps were closed by primer walking and sequencing of the PCR products. Any possible wrong assembly was corrected using PCR amplification and direct sequencing. Genes were predicted from the assembled result using Glimmer 3.02, which was developed for microorganisms. Tandem repeats were predicted using Tandem Repeat Finder 4.04. Mini- and microsatellite DNA were selected based on the number and length of the repeat units.
The rRNA database BLAST analysis was used to identify rRNA-encoding genes. rRNAmmer 1.2, tRNAscan 1.23, and Infernal software and Rfam 10.1 database were used to predict the rRNA, transfer RNA (tRNA), and small RNA (sRNA)-encoding genes, respectively (Lowe and Eddy, 1997; Lagesen et al., 2007; Gardner et al., 2009). The result of rRNA database BLAST search is precise but not complete, and therefore, rRNAmmer was used to predict rRNA genes when there was no homology reference. The final result is a combination of all the above methods.
The nucleotide sequence of 16S rDNA and P36 gene were pairwise aligned with ClustalW1.6, the Neighbor-Joining method using the p-distance to compute the evolutionary distances, and the pairwise deletion of gaps was implemented in the MEGA6.06 software program. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) is shown next to each branch (Siqueira et al., 2013; Gonchoroski et al., 2017). The cutoff value for condensed tree is 50.
The Open Reading Frame (ORF) is also known as Coding sequences. The ORF of the M. h TB1 strain was predicted by Glimemer 3.02. ORFs longer than 30 amino acids were selected, each ORF was searched for protein homology in the Non-Redundant Protein Sequence Database (the nonredundant protein), and the refseq_genomic (Reference genomic sequence) database by Blastp, comparing the sequence and target sequences to more than 30% specificity and more than 60% homologous sequences are considered to be significantly correlated, and the gene names and functions are annotated. We used tRNAscan-SE software to predict the transport of RNA-related genes in the genome. Finally, we used ARTEMIS to modify, collate, and visualize the gene predictions and annotations, and then submit the annotated sequences to NCBI by the Sequin software.
The predicted ORFs are annotated by Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Swiss-Prot, NR database, and Cluster of Orthologous Group of proteins (COG) database, respectively.
Protein sequences were analyzed for function annotation. The genes were aligned within GenBank databases to obtain their corresponding annotations by FGENESH. The result with the highest quality alignment was selected for gene annotation for biological relevance. Function annotation was completed by BLAST search of the genes with different databases, including the KEGG, COG, SwissProt, NR, and GO.
Results
Morphological observation of M. hyopneumoniae
The Mh pathogen isolated from the lungs of Tibetan pigs was characterized microscopically and its DNA was sequenced. The typical morphology of Mh was evident in Wright's and Giemsa-stained smears (Fig. 1). Blue globular bacteria was visible after the staining.
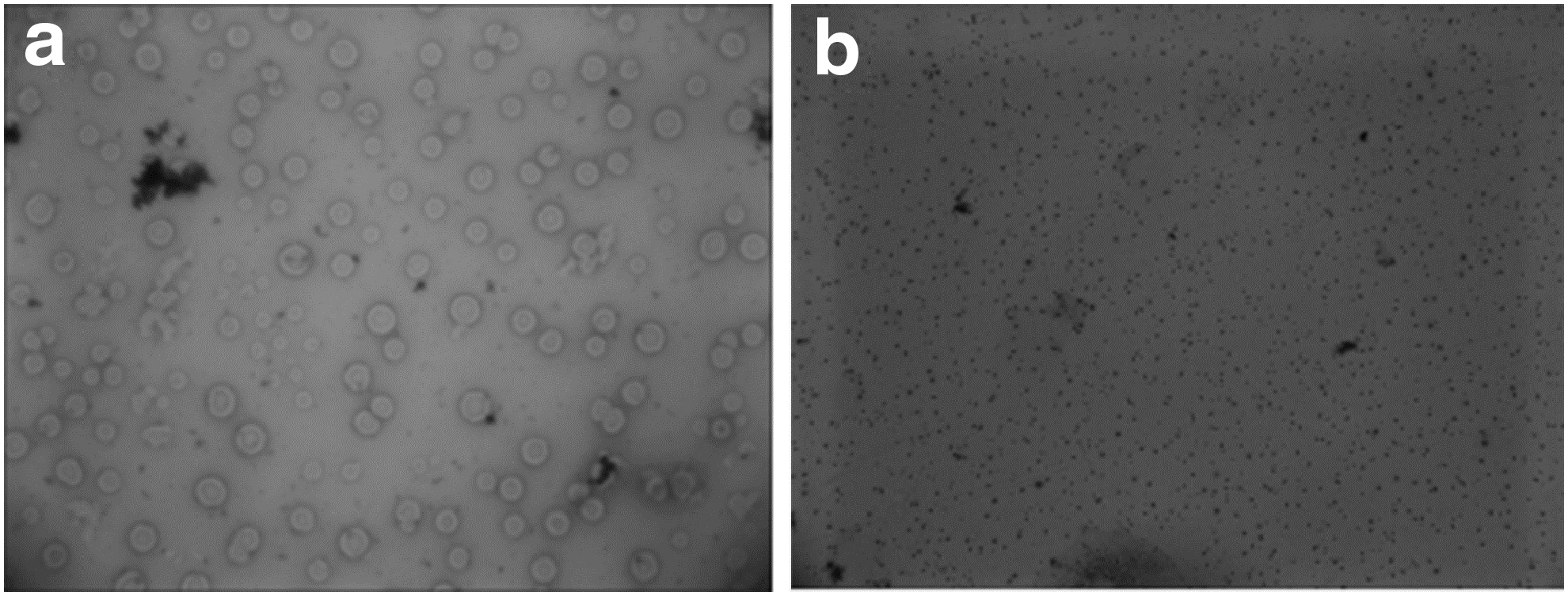
Morphological observation of Mycoplasma hyopneumoniae TB1 isolated from lungs of Tibetan pigs.
PCR analysis of the 16S rDNA and the species-specific gene P36 of M. hyopneumoniae TB1 isolated from Tibetan pigs
Gel images of the PCR-amplified 16S rRNA and P36 of M. hyopneumoniae are shown in Figures 2 and 3, respectively. Figure 4 displays a molecular phylogenetic tree based on the P36 gene sequences in the isolated samples.

PCR of the 16S rDNA of M. hyopneumoniae TB1 isolated from Tibetan pig lungs. Lanes: M: DL2000 DNA marker; P: positive control (Mh strain 7448 [GenBank: NC_007332]); N: negative control (double-distilled water); 51 − 58: experimental bacterial samples (eight successful isolates). 87, 94, 97: control group (vaccine strain RM48), 16S rDNA gene of number 51 strain (TB1) GenBank accession number: KY003100. 16S rDNA gene of number 52 strain (TB2) GenBank accession number: KY003101. PCR, polymerase chain reaction; rDNA, recombinant DNA.
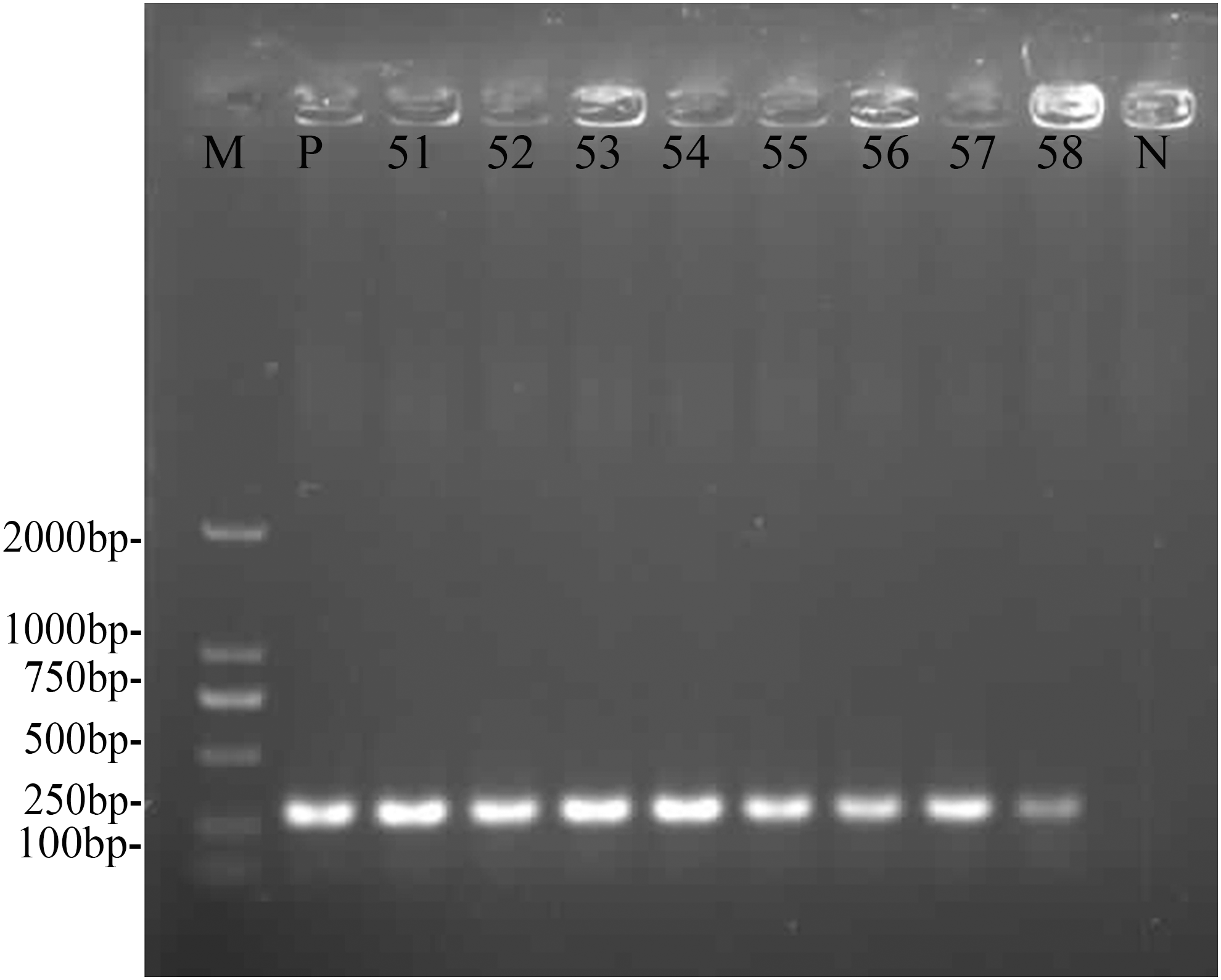
PCR of the species-specific gene P36 from M. hyopneumoniae TB1 isolated from Tibetan pig lungs. Lanes: M: DL2000 DNA marker; P: positive control; N: negative control; 51–58: experimental bacterial samples. P36 gene of number 51 strain GenBank accession number: KY290169.

Molecular phylogenetic tree of P36 of M. hyopneumoniae TB1.
Molecular phylogenetic and genomic analysis
Sequence alignment of the 16S rDNA or P36 and analysis of the sequences using NCBI BLAST indicated high sequence homology of the isolated bacteria with Mh. Phylogenetic characterization using the entire 16S rDNA sequences indicated close identity with Mh strains 7448 (GenBank: AE017244.1, 99%), 232 (GenBank: AE017332.1, 99%), 168 (GenBank: CP002274.1, 99%), J (GenBank: AE017243.1, 99%), 168-L (GenBank: CP003131.1), and 7422 (GenBank: CP003802.1, 99%). Comparable results (i.e., 99% identity) were obtained upon analyzing the P36 sequences. Therefore, the pathogenic microorganism isolated from Tibetan pig lungs was clearly identified as Mh, although distinct from strains 7448, 232, 168, J, 168-L, and 7422.
The library preparation method and sequencing pipeline is shown in Supplementary Figure S1. Sequencing statistics of Mh TB1 is shown in Supplementary Table S1).
K-mer analysis of M. hyopneumoniae TB1
Before assembling, k-mer analysis was used to estimate genome size, the degree of heterozygosis, and the degree of duplication. The genome size of the Mh TB1 strain is ∼0.65 mb (Supplementary Fig. S2). The assembled results of the Mh TB1 strain indicated a genome size of 909,064 bp, 28.53% GC content, 57 scaffolds, and 237 contigs (Supplementary Table S2).
GC content, depth correlation, and gene length distribution analysis of M. hyopneumoniae TB1
Gene prediction was executed for the assembled results, and the statistics of gene length was depicted in Supplementary Figure S3. GC distribution (Supplementary Fig. S4) was obtained by GC-Depth analysis. The number of tandem repeat sequence was 91, and the number of mini- and microsatellite DNA was 50 and 15, respectively (Supplementary Table S3). Prokaryotic rRNA contains 5S rRNA, 16S rRNA, and 23S rRNA. Generally, for the Mh TB1 strain, the 16S and 23S rRNA consisted of 1,540 and 2,900 nucleotides, respectively (Supplementary Table S4).
KEGG and COG annotation of M. hyopneumoniae TB1, M. hyopneumoniae TB1 genome architecture, and dilution curve analysis
The statistics of KEGG annotation (Fig. 5) could be used to provide an overview of the KEGG analysis results. COG annotation data are provided in Figure 6 and the Mh TB1 genome architecture is depicted in Figure 7. The genomes of the different Mh strains were compared. The genes shared by all Mh are core genes, which are essential for growth. Genes present in only one strain were considered as differential genes. Knowledge of differential and core genes is important for the detection of functional differences and similarities between samples, which provide molecular evidence for phenotype differences and similarities. In the core/pan gene analysis, the reference was Mh 168, and the queried strains were Mh J, Mh 7448, Mh 232, Mh 7422, and 2B (TB1 strain). The results are shown in Table 2 and Figure 8. The heatmap after core gene deletion is shown in Figure 9.
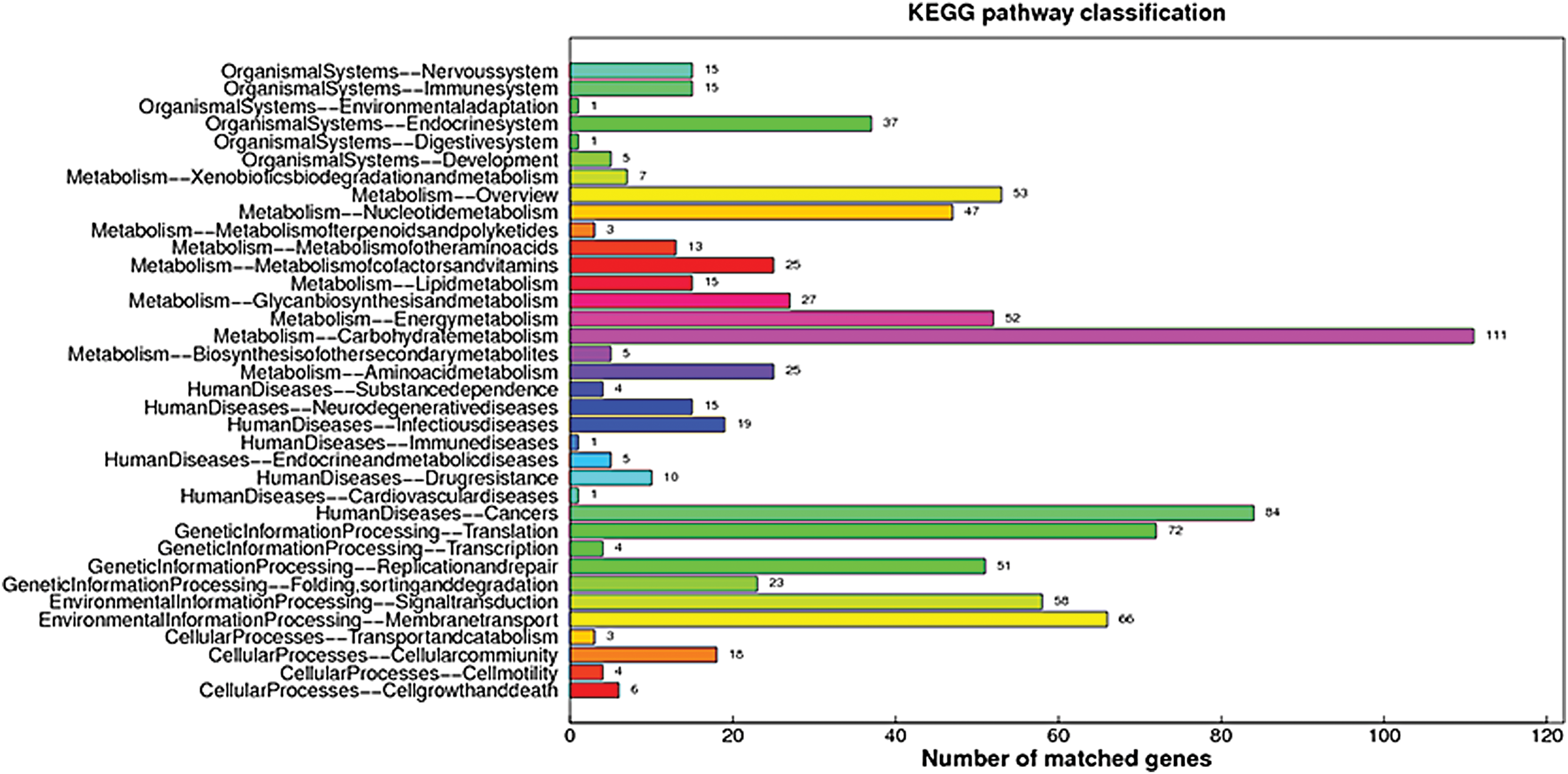
KEGG annotation of M. hyopneumoniae TB1. KEGG, Kyoto Encyclopedia of Genes and Genomes. Color images are available online.
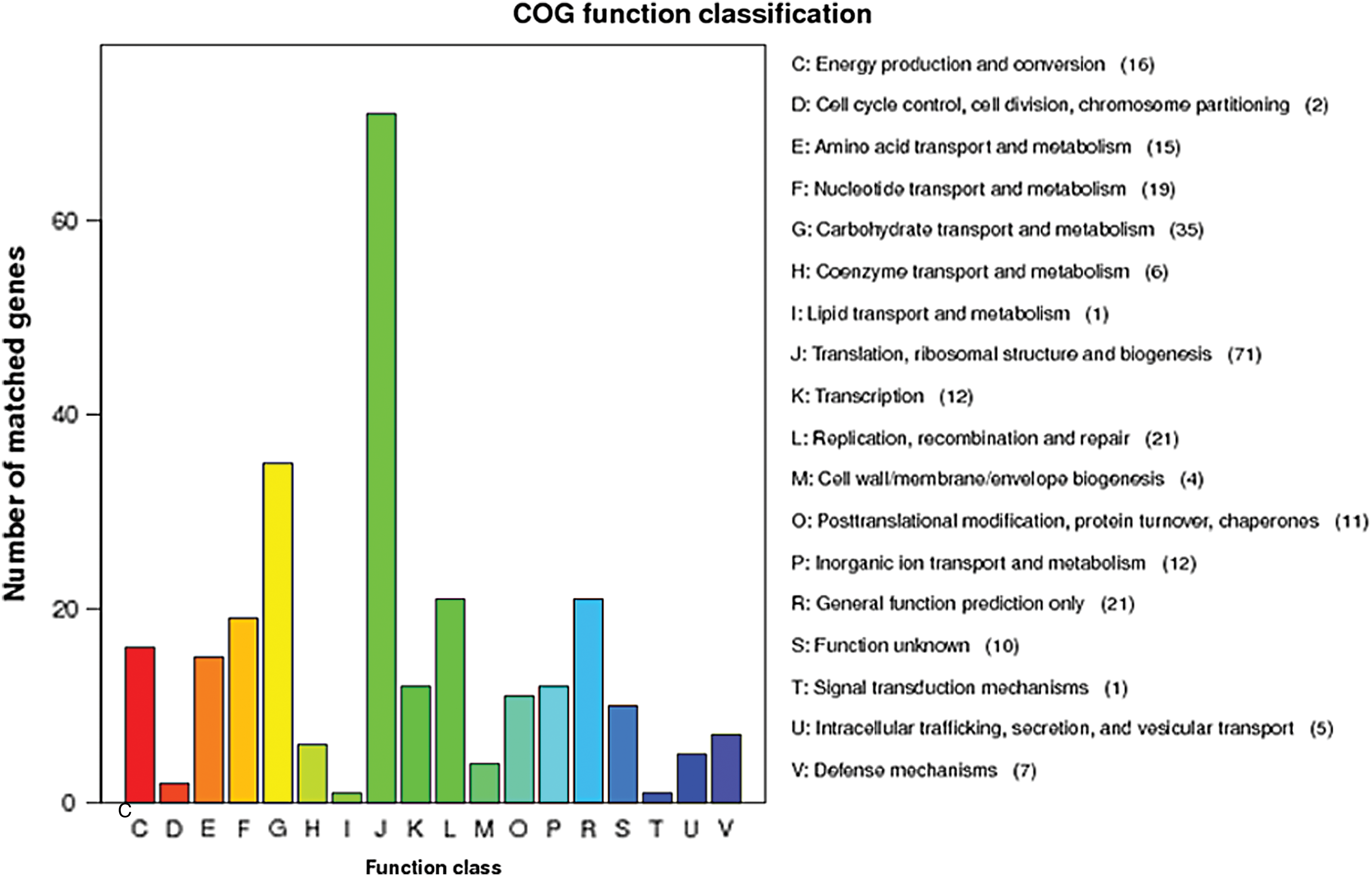
COG annotation of M. hyopneumoniae TB1. COG, Cluster of Orthologous Group of proteins. Color images are available online.

M. hyopneumoniae TB1 genome architecture. The dnaA gene is at position zero. Moving inside, the first circle shows the genome length (units in Mh); the second and the third circles show the locations of the predicted CDSs on the plus and minus strands, respectively, which are color-coded according to the COG categories (the color codes for the functional assignments are shown in the key). CDSs, coding sequences. Color images are available online.
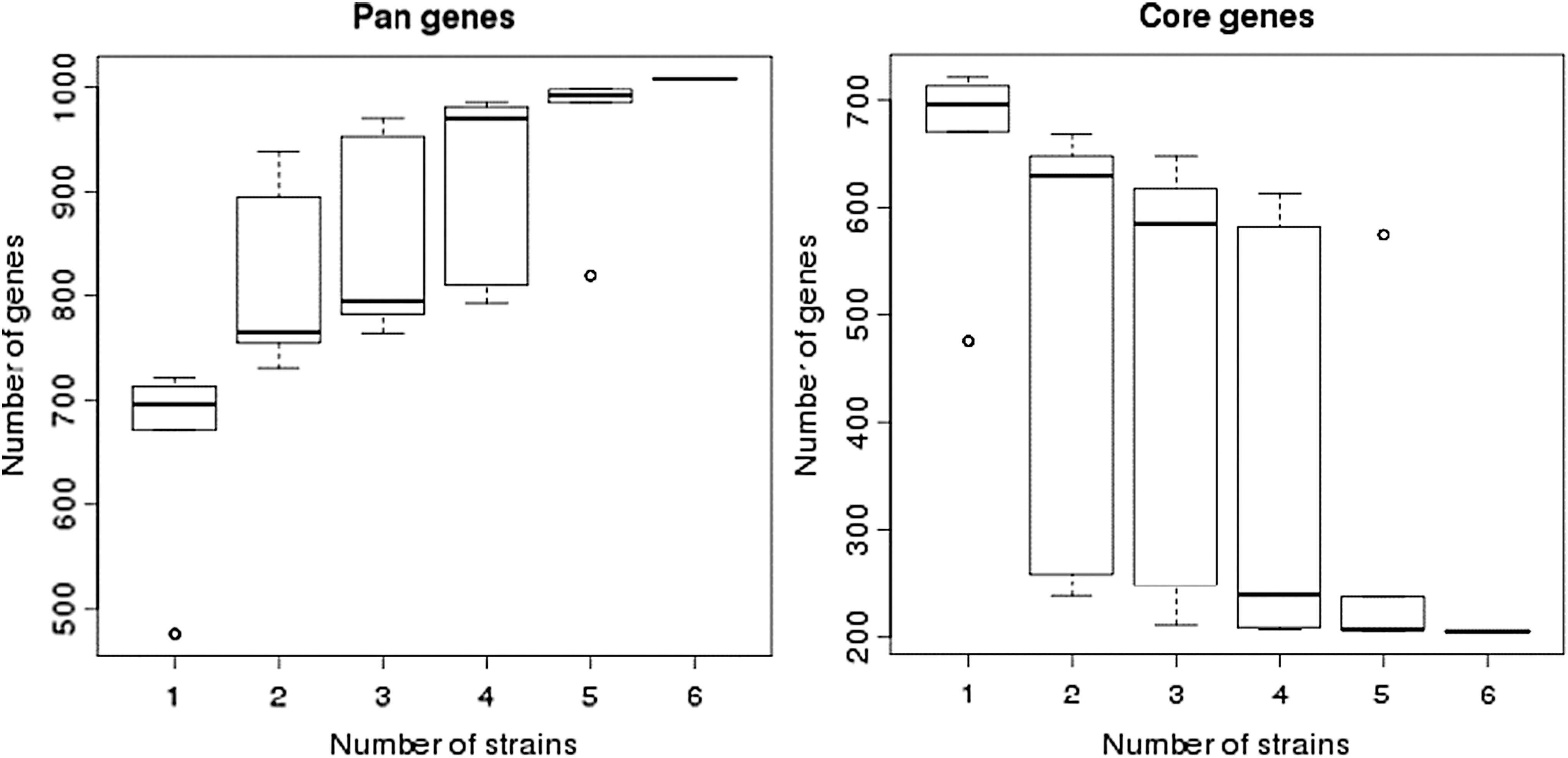
Dilution curve of M. hyopneumoniae TB1 genes. Core/pan gene analysis with references: 1, Mh 168, query; 2, Mh J; 3, Mh 7448; 4, Mh 232; 5, Mh sample B; 6, Mh 7422.
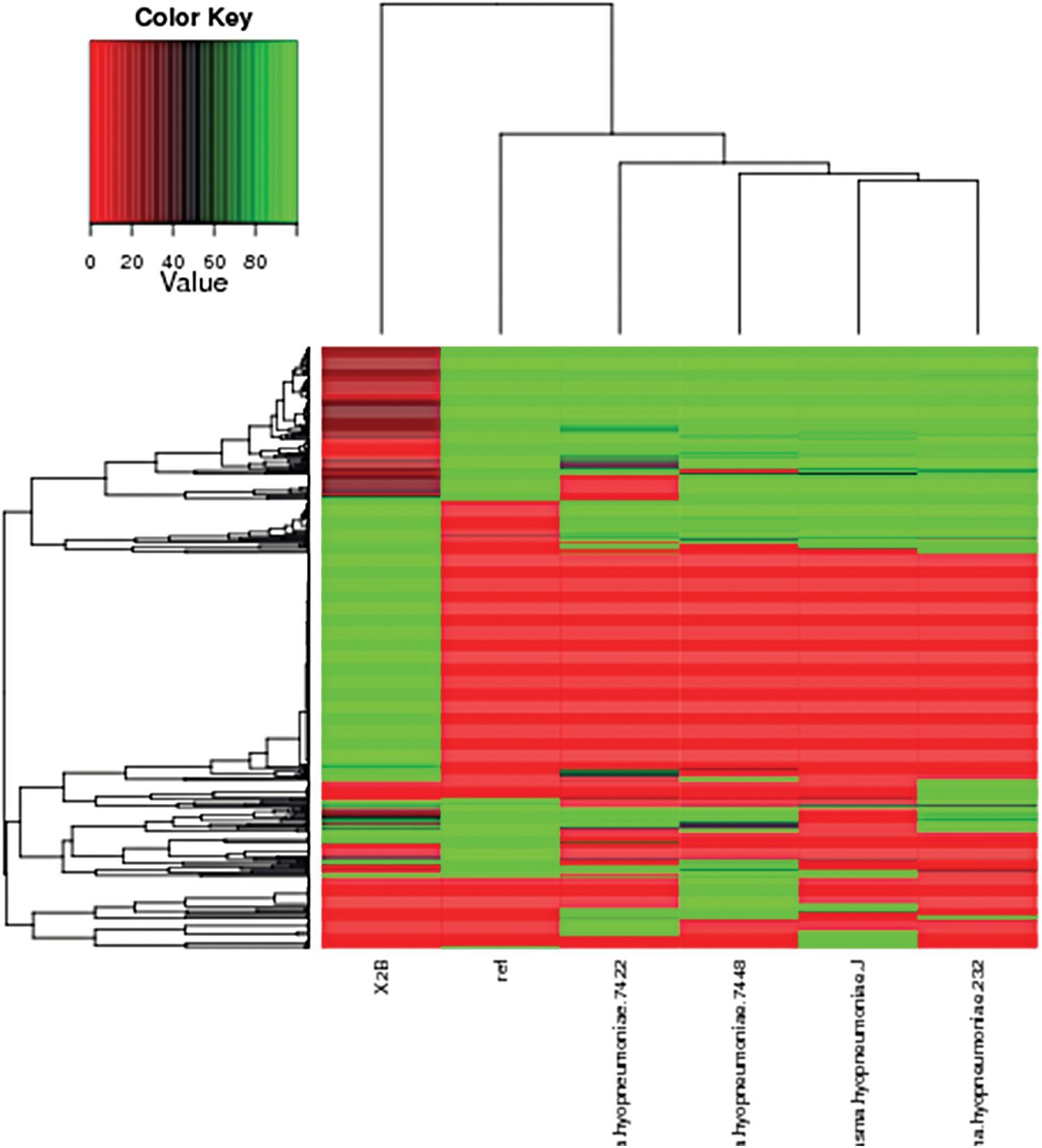
Heatmap after core gene deletion of M. hyopneumoniae TB1. Core/pan gene analysis with references: Mh 168, query: Mh J, Mh 7448, Mh 232, Mh 7422, Mh sample B (X2B). Color images are available online.
Number and Size of Core-Pan Genes
amino acids.
Phylogenetic tree construction using M. hyopneumoniae TB1 and reference strains
Partial sequence of 16S rDNA (PCR product, KY003100) of isolated strain and homologous gene in GenBank database were used for neighbor-joining BLAST analysis to construct the phylogenetic trees. The NCBI sequences of six Mh strains (M. hyopneumoniae 7448, 232, 168, 168-L, J, and 7422) were used for neighbor-joining BLAST analysis to construct the TB1 strain (NBBN00000000) phylogenetic trees. The 16S rRNA phylogenic tree and genome phylogenic tree of Mh TB1 are presented in Figures 10 and 11, respectively. The wild Mh genome displayed high synteny although partial mutation in the nucleic acid sequence of the wild Mh strain was evident when compared with the reference genomes of the five strains (J, 7448, 232, 7422, 168, and 168-L).

16S rDNA phylogenic tree of M. hyopneumoniae TB1 (KY003100).

Genome phylogenic tree of M. hyopneumoniae TB1 (NBBN00000000).
Annotations of the Virulence Factors of Pathogenic Bacteria (VFDB) database revealed virulence factors of the Mh TB1 strain, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH; MG_301), the molecular chaperone DnaK (CT396), an oligopeptide ABC transporter, a permease component (oppF), an ATP-dependent protease (clpE), a putative prolipoprotein p65 (p65), pyruvate dehydrogenase (lipoamide) e1-beta chain (pdhB), elongation factor Tu (tuf), lipoyltransferase and lipoate-protein ligase family protein (lplA1), phosphopyruvate hydratase (eno), membrane nuclease, lipoprotein (nuc), and hemolysin (hlyA).
Comparative genomic analyses revealed genome characteristics of Mh TB1 related to high-altitude hypoxia (plateau hypoxia) and unique virulence factors.
Discussion
Genomic analysis confirmed the identity of the bacteria isolated from porcine lungs as Mh and revealed the underlying pathogenic information. In general, Mh TB1 sequence is conserved, with its core gene being homologous to the reference strains. Owing to the extreme environment in the Tibetan plateau, the Mh TB1 genome showed differences in gene number, especially for differential genes. Significant changes were observed for genes involved in energy metabolism and virulence. It is speculated that M. hyopneumoniae in Tibetan pigs enriches our understanding of M. hyopneumoniae and expands genetic diversity. Further understanding of the diversity of M. hyopneumoniae genes in Tibetan pigs is one of the next steps.
Successful isolation of this pathogen is considered the gold standard for diagnosis of Mh infection in pigs (Marois et al., 2010). In Tibet, acquisition of pig lungs and contamination with other bacteria are the two main limiting factors in this procedure. The Mh isolation requires longer bacterial culture time than Escherichia coli, but the success rate of separation is much lower than E. coli. However, as far as we know, this report is the first to describe the isolation and identification of this pathogen from Tibetan pigs and provide a genomic comparison with reference strains.
The isolation rate of Mh TB1 was only 6.7% from lung tissue of pigs that showed typical pathological changes (8 successful isolations from 120 samples). The low separation rate reported in this study is consistent with previous reports. Maurício et al. (2015) reported a separation rate of 1.70% from pigs. In comparison, as the local breed, it is inferenced that the infection rate of Mh to the Tibetan pigs is higher. The rate of successful Mh isolation depended on various factors. Strictly aseptic manipulation and use of 0.22-μm microporous membrane filtration were prerequisites for successful separation. Different batches of phenol red differentially affected the culture. Acetic acid thallium (0.25%) and cycloserine, penicillin, ampicillin, or flucloxacillin, alone or in combinations, inhibited growth of fungal and gram-positive bacterial contaminants when added to the culture. The methods of tissue treatment, cutting, and homogenization were important for the effective isolation of Mh. In particular, in a prior study, the outer surface of the lung was soaked in boiling water for 2 s (Guimaraes et al., 2014) and the samples were cut from the junction of diseased and healthy tissue. Diseased materials must be refrigerated or immersed in penicillin G or ampicillin-containing liquid medium during transportation to the laboratory (Abe et al., 2016).
GAPDH is a 37 kDa enzyme that catalyzes the sixth step of glycolysis, which is used to decompose glucose to produce energy. In addition to this important metabolic function, GAPDH is involved in a variety of nonmetabolic processes, including initiation of apoptosis, transcriptional activation, endoplasmic reticulum to Golgi vesicle bypass, and rapid axonal or axonal transport.
Molecular chaperones of the 70 kDa family mediate protein/protein interactions by selectively binding to partially unfolded segments of other proteins in an ATP-dependent activity cycle. Previous investigations of chaperone substrate selectivity have shown that chaperones have a propensity to bind to partially unfolded segments of polypeptides that contain bulky hydrophobic residues. Recent investigations have shown that 70 kDa chaperones, such as DnaK, which is expressed by E. coli, also bind short basic peptides and even polycations (Witt et al., 1997).
Putative oligopeptide permease ABC transport operon (opp) consists of five genes (oppB, oppC, oppD, oppF, and oppA), encoding two permeases, two ATPases, and a substrate-binding protein. Opp system mediates both uptake of peptides and fitness in the respiratory tract (Jones et al., 2014).
ClpE is required for the ability of bacterial surviving, adhering, and invading to host cells, and it plays an important role in the pathopoiesis of Mh.
RELA, also known as p65, is a REL-associated protein involved in NF-κB heterodimer formation, nuclear translocation, and activation.
Elongation factor Tu (tuf) mediates the entry of the aminoacyl tRNA into a free site of the ribosome. Elongation factors are also a target for pathogen toxins. Lipoate-protein ligase (LplA1) is a cofactor required for the function of alpha-keto acid dehydrogenase and glycine cleavage enzyme complexes. The naturally occurring form of lipoate is attached by amide linkage to the epsilon-amino group of a specific lysine residue within conserved lipoate-accepting protein domains. Lipoate-protein ligase(s) catalyze the formation of this amide bond between lipoyl groups and specific apoproteins (Morris et al., 1994).
Membrane nucleases of mycoplasmas are believed to play important roles in growth and pathogenesis (Jarvill-Taylor et al., 1999).
Lipoproteins of the Mh cell membrane surface play a pivotal role in pathogen–host interactions, antigenic variation, and immunity evasion. Lipoproteins are considered to be responsible for Mycoplasma virulence (Bürki et al., 2015). Aligned by VFDB database, lipoprotein (nuc) was found. Lipoprotein (nuc) could be considered as pivotal virulence factors.
Hemolysins are toxic proteins that attack erythrocyte membranes and cause cell rupture (Chen et al., 2019).
Future studies will examine whether the eight isolates (samples 51–58) are 100% identical or whether variations are present (Mayor et al., 2007). We conclude that the present outbreak of pig lung disease in Tibet is associated with Mh. This study demonstrates that M. hyopneumoniae continues to circulate among Tibetan pig herds in the Qinghai-Tibetan Plateau and might be underdiagnosed. Thus, flock monitoring for Mh is critical. Genomic analysis revealed comparable results between strain TB1 and the reference strains. In general, Mh is genetically conserved, although differential genes are present and were detected in greater prevalence than previously reported. We hypothesize that the genetic variation among the isolated strains may be due to long-term extreme natural selection pressure (Xia et al., 2017). Mh disseminates through air, and hence, it must be tenacious enough to withstand damage by the plateau environment, which includes high-altitude hypoxia and low pressure, low air humidity, and intense ultraviolet exposure. This study depends on the existing essay, the results based on a small sample size and may have limited generalizability. The next step is to explore the mechanism of the plateau adaptation and analyze its pathogenic mechanism.
Footnotes
Acknowledgments
This work was supported by the Henan Province Science and Technology Department Industry–University–Research Cooperation Project, “Study on the Prevention and Treatment of Porcine Respiratory Syndrome (PRDC) by Probiotics Fermentation Xinyang genuine Chinese Herbal Medicine Ranunculus ternatus Thunb” (Project No. 172107000045), the joint fund of the Tibet Autonomous Region, Department of Science and Tibet Agriculture and Animal Husbandry College ([Project No. 2016ZR-NQ-4] titled “Molecular epidemiology of Mycoplasma hyopneumoniae in Tibetan pigs,” and the Tibet Agriculture and Animal Husbandry College students' innovative experimental training program [Project No. 201801]) titled “Preliminary development of inactivated vaccine against Mycoplasma hyopneumoniae of Tibetan pigs.”
Author Contributions
G.Q. designed the study. G.Q., Z.Q.H., X.L., S.H., and L.Z. collected samples. G.Q. and Y.R. performed the experiments. G.Q., B.Y., T.L., and K.L. analyzed the data. G.Q. wrote the article and carefully revised the article. All authors read and approved the final version of article.
Disclosure Statement
The authors declare that they have no competing interests.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.