
Abstract
Dilated cardiomyopathy (DCM) is a common type of cardiomyopathy. The pathogenesis of DCM remains unclear and involves varied genes. AXIN1 is a crucial gene in regulating various functions in cells, it encodes protein Axin1, which regulates the assembly and disassembly of β-catenin destruction complex. In addition, Wnt/β-catenin signaling pathway plays an important role in cardiogenesis. We aimed to detect whether AXIN1 polymorphisms contribute to the susceptibility and prognosis of DCM in a Chinese Han population. A total of 340 DCM patients and 430 controls were enrolled, and patients who had complete contact information were followed up for a median period of 49 months. Polymerase chain reaction-restriction fragment length polymorphism was carried out to genotype the two AXIN1 tag single nucleotide polymorphisms (SNPs) (rs12921862 and rs1805105). All data were analyzed using the statistical software package, SPSS 21.0. The frequencies of allele A in rs12921862 and allele C in rs1805015 were increased in DCM patients compared with healthy controls (p < 0.001). Genotypic frequencies of rs12921862 and rs1805105 were associated with the susceptibility of DCM in codominant, dominant, and overdominant models (p < 0.01). AA/AC and AC genotypes of rs12921862 in the dominant and the overdominant genetic models also presented a correlation with poor prognosis of DCM in both univariate (p < 0.01) and multivariate analyses (p < 0.01) after adjusting for age, gender, left ventricular (LV) end-diastolic diameter, and LV ejection fraction. Our results suggest that AXIN1 polymorphisms are associated with the susceptibility and prognosis of DCM in a Chinese Han population.
Introduction
Dilated cardiomyopathy (DCM) is defined by the presence of left ventricular (LV) dilatation and LV systolic dysfunction, whereas hypertension, valve disease, or coronary artery disease are excluded from the etiology diagnosis criterion (Elliott et al., 2008). DCM is an irreversible and a general type of cardiomyopathy with a prevalence of 1 in 2500 individuals (Taylor et al., 2006). It is one of the critical causes of heart failure, and the most common indication for heart transplantation (Maron et al., 2006). The causes of DCM are varied, including genetic and nongenetic factors, with the genetic causes playing an important role at all ages. More than 50 disease-related genes that are involved with cytoskeletal, sarcomere, and nuclear envelope proteins have been reported (Morales and Hershberger, 2013; Pinto et al., 2016). These genes account for up to 35% of cases, and among all DCM cases, 20–35% are made up of familial DCM (Michels et al., 1992; Pinto et al., 2016; Weintraub et al., 2017). These findings suggest that genetic components are tightly correlated with the occurrence and development of DCM.
As reported, the Wnt/β-catenin signaling pathway plays a pivotal role during embryonic development, as it controls cell proliferation, differentiation, stem cell maintenance, and adult tissue homeostasis. Aberration of this pathway can cause a variety of human cancers, human birth defects, and other diseases (Clevers, 2006; Song et al., 2014; Wang et al., 2018). Moreover, the Wnt/β-catenin signaling pathway is activated in response to cardiac injury or stress in adult mammals, and facilitates cardiac hypertrophy and remodeling (Bergmann, 2010). Several studies have delineated that the canonical Wnt/β-catenin signaling pathway regulates cardiac development in a biphasic manner: it promotes cardiogenesis at an early stage but inhibits cardiac specification later on (Naito et al., 2006; Ueno et al., 2007).
As a downstream effector molecule, β-catenin is a key factor in the regulation of the canonical Wnt/β-catenin signaling pathway (Ozhan and Weidinger, 2015). However, it can be phosphorylated, ubiquitinated, and subsequently degradated by a dedicated cytoplasmic destruction complex. This complex is composed of a central scaffold protein named Axin, adenomatous polyposis coli, kinases glycogen synthase kinase-3 alpha/beta (GSK-3), and casein kinase-1 (CKI) (Li et al., 2012). Axin1 is the rate-limiting factor of the β-catenin destruction complex, and it regulates the complex's assembly and disassembly rapidly (Lee et al., 2003), whereas genetic changes in the components of the destruction complex can lead to human cancers (Najdi et al., 2011).
Variation of AXIN1 has been examined in many diseases, such as colorectal cancer, breast cancer, and cryptorchidism (Parveen et al., 2011; Zhou et al., 2015; Chimge et al., 2017), and the association between AXIN1 polymorphisms and atrial septal defect has been demonstrated in a previous study (Pu et al., 2014). Thus, we assumed that AXIN1 may be correlated with the susceptibility and prognosis of DCM. To verify this hypothesis, we selected two tag SNPs (rs12921862 and rs1805105), which are located in the intron and exon regions of AXIN1, respectively, and investigated the role of AXIN1 in DCM in a Chinese Han population.
Materials and Methods
Study subjects
In this case–control study, a total of 340 unrelated DCM patients (mean age: 50.3 ± 14.0; gender: male, 216; female, 124) were recruited between June 2002 and July 2016 from West China Hospital of Sichuan University. The diagnosis of DCM was made consistent with criteria established by the World Health Organization/International Society and Federation of Cardiology Task Force on the Classification of Cardiomyopathies in 1995 (before 2006) and the scientific Statement on the Definitions and Classification of Cardiomyopathies proposed by the American Heart Association in 2006 (after 2006) (Richardson et al., 1996; Maron et al., 2006). Clinical data were gleaned from the hospital record system.
A total of 430 healthy individuals (mean age: 50.8 ± 6.6; gender: male, 225; female, 205) from routine health surveys were enrolled as a control group. Patients with a history of hypertension, cardiac valve disease, coronary artery disease, congenital heart disease, tachyarrhythmia, pericardial disease, heavy alcohol intake, acute viral myocarditis, systemic diseases of a putative autoimmune origin, skeletal myopathies, diabetes, and nutrition disorders were excluded. Individuals of the control group had no evidence of organic cardiac disease and cardiac dysfunction, and their echocardiogram results were normal. All subjects were from the Han population living in southwestern China. This study was performed with approval from the West China Hospital Ethics Committee.
SNP selection and genotyping
Given that linkage disequilibrium extensively exists in genetic patterns, we selected two tag SNPs of AXIN1, based on the data of the Han Chinese in Beijing China (CHB) population sample of the HapMap Project (Data Release 24/Phase II, NCBI build 36 assembly, dpSNPb126). Eventually, rs12921862 and rs1805105 were picked out using the algorithm Tagger pairwise Tagging from the international HapMap project (
Two milliliters of peripheral venous blood was drawn from all subjects with informed consent, and genomic DNA was isolated from 200 μL of the EDTA-anticoagulated peripheral blood samples using a commercial DNA isolation kit (BioTeke, Peking, China) following manufacturer's instructions.
Genotyping of the two AXIN1 SNPs (rs12921862 and rs1805105) that were selected was performed using polymerase chain reaction restriction fragment length polymorphism (PCR-RFLP). The amplification reacted in a 10 μL mixture, containing 0.6 μL of genomic DNA, 5 μL of Power Taq PCR Master Mix (BioTeke), 0.15 μL each of a forward and a reverse primer, and 4.1 μL ddH2O. PCR was performed under the following conditions: 94°C for 4 min, followed by 34 cycles of denaturation at 94°C for 30 s, annealing at 56°C (rs12921862) and 66°C (rs1805105) for 30 s, extension at 72°C for 30 s, and final elongation at 72°C for 10 min. Subsequently, 1 μL of the PCR products was digested within a 10 μL restriction enzymes reaction solution at 37°C for 2 h (scrFI for rs12921862) or 1 h (FokI for rs1805105) (New England Biolabs, Peking, China) (Table 1). The digested products were visualized using 6% polyacrylamide gels with silver staining. To confirm the genotypes, the mentioned procedures were randomly repeated on ∼10% of the samples, and results were found to be concordant.
Primer Sequences and Polymerase Chain Reaction-Restriction Fragment Length Polymorphism Conditions for Two SNPs in AXIN1 Gene
Clinical follow-up
A total of 155 patients who had complete contact information were followed up for every 3 months, and the endpoint was cardiac death or loss of follow-up. A double-blind method was performed during the whole process regarding the genotype of the patient. Baseline data (gender and age) and echocardiographic indicators (LV end-diastolic diameter [LVEDD], and LV ejection fraction [LVEF]) were obtained from medical records and echocardiography was performed at the time of diagnosis.
Statistical analysis
Data analyses were conducted using the statistical software package SPSS 21.0 (SPSS, Inc., Chicago, IL). Shapiro–Wilk's test was used to determine normality. For continuous data, the Mann–Whitney U test was performed to analyze the differences between two dependent samples, and categorical data were compared using Pearson's chi-square test. “Power and Sample Size Calculation” software (version 3.0.43) was used to detect sample power (Hong and Park, 2012). Genotypic and allelic frequencies were directly obtained by counting the number. Associations between two SNPs and susceptibility to DCM were evaluated in the codominant, dominant, recessive, and overdominant models of the SNPstats website, and the Hardy–Weinberg equilibrium (HWE) of the control group was calculated using the chi-square test. The odds ratio (OR) and its 95% confidence intervals (95% CIs) were obtained to evaluate the effects of any differences between alleles or genotypes. Meanwhile, survival analyses were done by using Kaplan–Meier and Cox regression analyses. A p value of <0.05 was considered statistically significant.
Results
Study subjects
According to the baseline data of the DCM patients, the DCM group had higher LVEDD (p < 0.001) and lower LVEF (p < 0.001) than the control group. All samples had >80% power in examining the association between AXIN1 polymorphisms and the risk of DCM.
Distribution of AXIN1 SNPs in DCM patients and patient characteristics and association with DCM susceptibility
The distribution of AXIN1 rs12921862 and rs1805105 genotype frequencies was in accordance with HWE (p > 0.05). To verify the association between the two SNPs polymorphisms and susceptibility to DCM, genotypic and allelic frequencies were compared between DCM patients and controls. Table 2 demonstrates significant differences with regard to both SNPs.
Distributions of AXIN1 SNPs in Dilated Cardiomyopathy Patients and Controls and Their Associations with Dilated Cardiomyopathy Risk
Bold values indicate statistical significance.
Adjusted for gender, age, LVEDD, and LVEF.
CI, confidence interval; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; OR, odds ratio.
For rs12921862, the frequency of allele A was 23.8% in DCM patients and 5% in healthy controls, which shows notable differences between the two groups (OR = 5.94, 95% CI = 4.17–8.46, p < 0.001). A significantly increased risk of DCM was found to be related with the AC genotype in the codominant model compared with that of the CC genotype (OR = 6.77, 95% CI = 2.52–18.17, p < 0.001). Furthermore, A allele carriers (AC/AA) were remarkably associated with DCM risk in the dominant genetic model (OR = 6.46, 95% CI = 2.49–16.77, p < 0.001). In the overdominant model, the frequency of the AC genotype was also higher in DCM than that in the control (37.1% vs. 8.6%, OR = 6.54, 95% CI = 2.43–17.59, p < 0.001).
For rs1805105, allele T frequency in DCM cases was lower than that in controls (57.9% vs. 77.7%, OR = 0.40, 95% CI = 0.32–0.49, p < 0.001). In the codominant model, the CT genotype presented a stronger correlation with susceptibility of DCM than the TT genotype (OR = 4.07, 95% CI = 1.79–9.26, p = 0.0026). Moreover, compared with the TT genotype, an increased risk of DCM was noted to be associated with the C allele carriers (CT/CC) in the dominant model (OR = 3.57, 95% CI = 1.61–7.94, p = 0.0013). In the overdominant model, the differences were also statistically significant since the frequency of the CT genotype was 68.8% and 31.6% in cases and controls, respectively (OR = 3.83, 95% CI = 1.75–8.41, p < 0.001).
There were no obvious differences between DCM patients and healthy controls in the recessive model for both rs12921862 (OR = 2.59, 95% CI = 0.16–42.28, p = 0.5) and rs1805105 (OR = 0.75, 95% CI = 0.15–3.61, p = 0.71). Hence, stratified analyses were conducted for this model. As shown in Table 3, after adjusting for age, LVEF, and LVEDD, the frequency of the AA genotype was higher in females than in males for rs12921862 of AXIN1 (8.9% vs. 3.2%, OR = 3.16, 95% CI = 1.16–8.63, p = 0.022). No correlation was found between gender and rs1805105, and there were no significant differences between the two SNPs of AXIN1 and the other indicators (age, LVEF, and LVEDD).
Distributions of AXIN1 SNPs in Patients' Characteristics and Their Associations with Dilated Cardiomyopathy Risk
Adjusted for age, gender, LVEDD, and LVEF.
Bold values indicate statistical significance
Association between AXIN1 SNPs and overall survival in DCM patients
Survival analyses were performed to determine the correlation between the two SNPs (rs12921862 and rs1805105) of AXIN1 and prognosis of DCM. A total of 155 patients (mean age: 50.1 ± 12.9; gender: male, 106; female, 49), for whom contact information was available, were followed up for a median period of 49 months. During this time, 25 patients were lost, and all included patients accepted continuous treatment and no one underwent heart transplantation. Of these patients, 91 were dead and 39 were alive at the end of the follow-up period. The results of the survival analyses are presented as Kaplan–Meier curves. AA/AC and AC genotypes of rs12921862 are associated with a worse prognosis of DCM patients in the dominant (log-rank: p < 0.001, Fig. 1) and the overdominant models (log-rank: p = 0.004, Fig. 2).
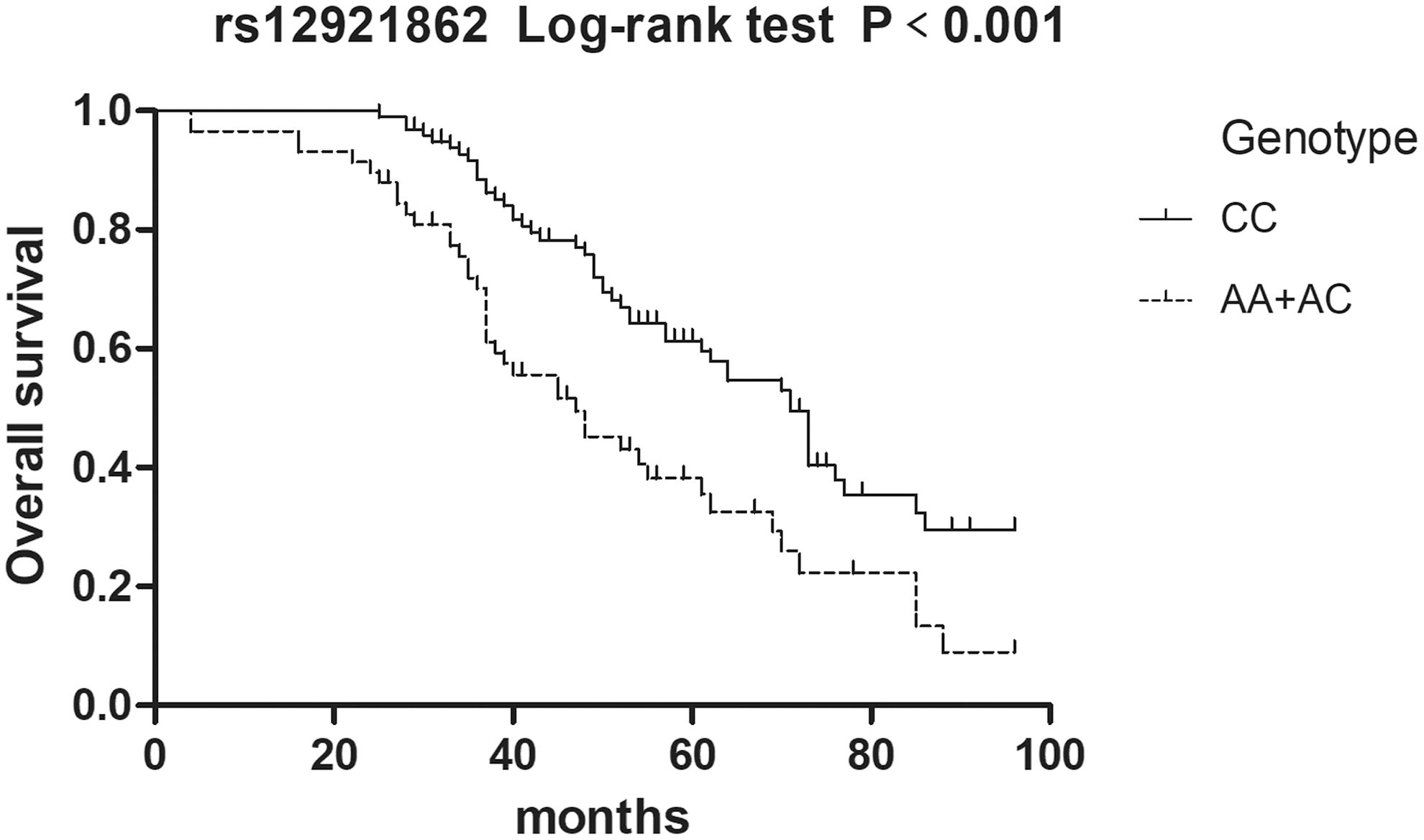
AA and AC genotypes of rs12921862 were associated with the worse prognosis of DCM patients in the dominant model (log-rank: p < 0.001). DCM, dilated cardiomyopathy.
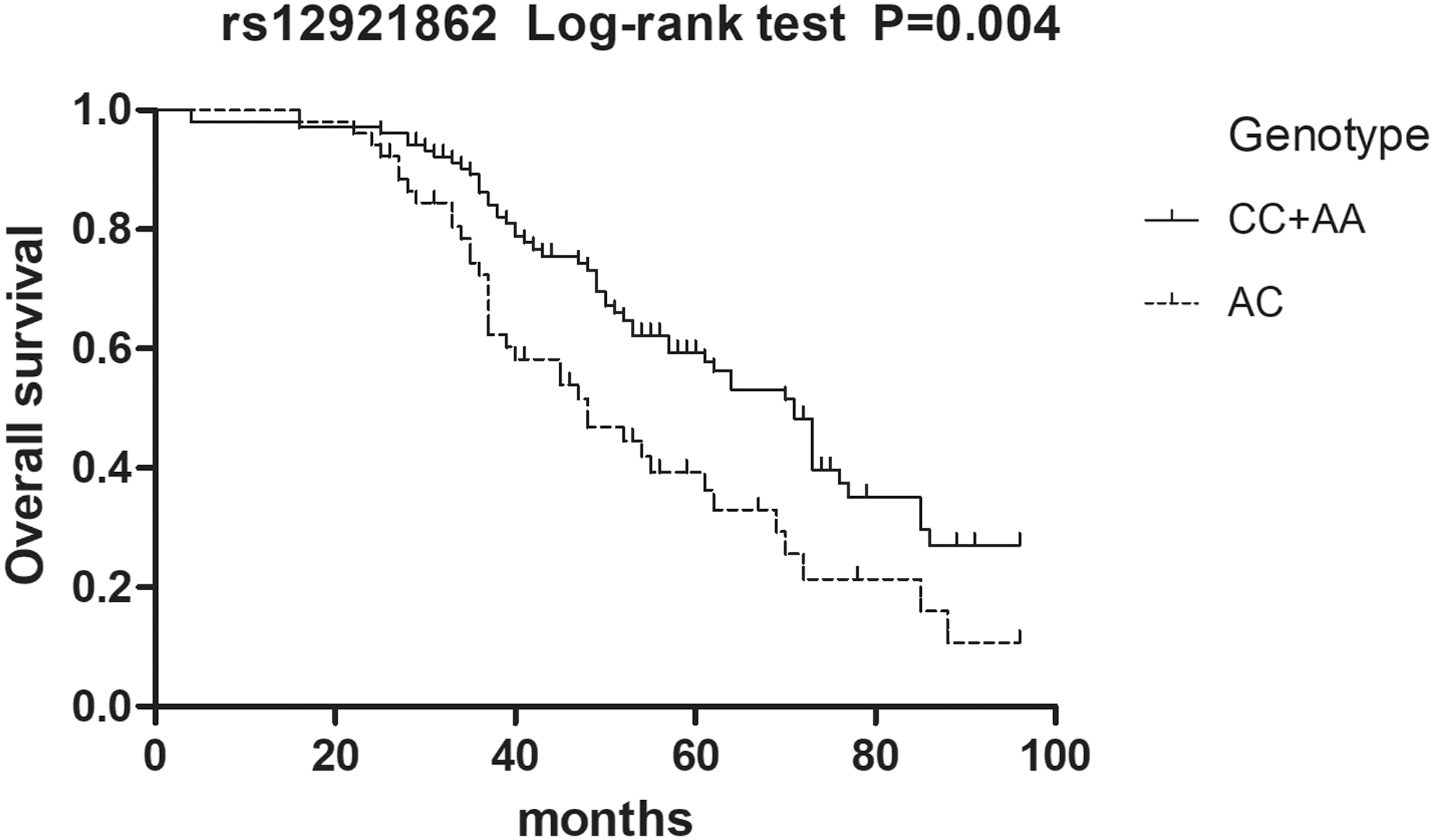
AC genotype of rs12921862 was associated with the worse prognosis of DCM patients in the overdominant model (log-rank: p = 0.004).
In the univariate Cox regression analysis, a positive correlation was found between the genotypic frequencies of rs12921862 and overall survival(os) of DCM patients in the dominant model (HR = 2.08, 95% CI = 1.37–3.16, p = 0.001), and the overdominant model (HR = 1.84, 95% CI = 1.21–2.82, p = 0.005), with the AA/AC genotypes, as well as the AC genotype in the overdominant model, indicating a worse prognosis. After adjusting for age, gender, LVEDD and LVEF, the results still remained statistically significant in the multivariate Cox regression analysis. No correlation was noted between rs1805105 of AXIN1 and the OS of DCM patients in the univariate and multivariate Cox regression analyses (Table 4).
Association Between AXIN1 SNPs and Overall Survival in Dilated Cardiomyopathy Patients
Bold values indicate statistical significance.
Adjusted for gender, age, LVEDD, and LVEF.
HR, hazard ratio.
Discussion
As previously reported, varied genes have been reported to be associated with the onset and development of DCM. However, since these genes encoding proteins are involved in extensive cellular functions, the pathological mechanisms that lead to DCM are diverse (McNally and Mestroni, 2017), and it is, therefore, arduous to determine the exact gene that is responsible for DCM on clinical grounds.
Most DCMs are inherited in an autosomal dominant pattern, with variable expressivity and age-dependent penetrance, especially in adulthood. Moreover, autosomal recessive, X-linked, and mitochondrial mutation inheritance have been reported to occur in DCM, which were more common in children than in adults (Hershberger et al., 2010, 2013; McNally et al., 2013). De novo variants can be seen in both children and adults (Hershberger et al., 2018). Although no precise evidence of penetrance has been reported, a previous study showed 90% penetrance after 40 years in AD forms (Mestroni et al., 1999). Thus, on account of the complexity of genetic inheritance and the high mortality of DCM, a better understanding of the pathogenesis is indispensable for further studies in DCM.
AXIN1 is located on chromosome 16p13.3, which has two isoforms, “isoform a” and “isoform b.” “Isoform a” encodes an 862aa polypeptide, whereas “isoform b” lacks the 36aa in the N-terminal domain encoded by exon 8 (Salahshor and Woodgett, 2005). As previously described, AXIN1 can regulate the Wnt/β-catenin signaling pathway by degradating β-catenin, with AXIN1 acting as a negative effector of the pathway (Willert et al., 1999).
β-Catenin is a multifunctional protein that can be phosphorylated and degradated by a destruction complex without the existence of Wnt. Nevertheless, in the presence of Wnt, β-catenin can accumulate in the cytoplasm, as Wnt binds to its receptor complex and results in the ablation of Axin (Gessert and Kuhl, 2010).
Previous studies have noted that the canonical Wnt/β-catenin signaling pathway regulates the proliferation and differentiation of embryonic cardiac precursors into mature cardiomyocytes (Ueno et al., 2007). Upregulation of Wnt/β-catenin is required at day 5.5 of the embryonic stage to ensure the proliferation of cardiac progenitor cell (CPC) pools. Subsequently, at day 7.5 of the embryonic stage, the downregulation of Wnt/β-catenin promotes the differentiation of the CPC pools into adult cardiomyocytes (Bergmann, 2010). Although this biphasic model appears to explain the function of Wnt/β-catenin in cardiogenesis, Susanne et al. summarized a model with four phases of cardiac specification and differentiation within the Wnt/β-catenin signaling pathway: first of all, the Wnt/β-catenin signaling pathway is required for the formation of the mesoderm and the endoderm; next, the pathway is downregulated for cardiac differentiation and specification of cardiac multipotent progenitor cells; then the Wnt/β-catenin signaling pathway is required again for the proliferation and expansion of certain CPCs; finally, Wnt/β-catenin signaling is downregulated for terminal differentiation (Gessert and Kuhl, 2010).
Disheveled (DVL) is an intracellular protein that can bind to the C-terminal region of Axin and inhibit Axin activation. Moreover, DVL can also bind to CKI, resulting in DVL phosphorylation, with phosphorylated DVL having a high affinity to Frat, a molecule that can bind to and inhibit GSK-3 (Kishida et al., 1999, 2001; Julius et al., 2000). Thus, the destruction complex is blocked, leading to the accumulation of β-catenin in the cytoplasm. Previous work has also indicated that the overexpression of DVL results in cardiac hypertrophy, LV enlargement, and reduction in LV ejection. However, cardiac hypertrophy induced by β-adrenergic can be abrogated by DVL knockdown cardiomyocytes (Malekar et al., 2010). Similarly, on pressure overload, deletion of β-catenin contributes to the attenuation of LV remodeling and the improvement of LV function, whereas in contrast, the expansion of LV and an increase in mortality were found in the β-catenin overexpression model (Baurand et al., 2007; Zhou et al., 2007; Zelarayan et al., 2008).
Although the two SNPs of AXIN1 are located in the intron and synonymous codon region, they have a dramatic effect on their mRNA secondary structure (Pu et al., 2017), which may cause aberrant gene regulation and phenotypic effect (Sabarinathan et al., 2013). In addition, the previous study suggested that introns and noncoding RNAs function as endogenous network control molecules, which enable direct gene–gene communication and multitasking of eukaryotic genomes (Mattick and Gagen, 2001). Taken together, these inferences indicate the feasibility of the role of AXIN1 in cardiogenesis and myocardiac remodeling, as well as in DCM pathogenesis. The two SNPs may function through regulating the interaction between genes rather than affecting the amino acid coding of the protein.
Our study is the first to investigate the association between AXIN1 and DCM in a Chinese Han population. In this study, the higher frequency of allele A in rs12921862 implies that allele A is a risk factor of DCM, as well as allele C in rs1805105. The number of male patients in DCM was 1.7-fold of the number of female patients, which is in accordance with the prevalence of DCM found in an earlier study by Petretta et al. (2011). Nevertheless, in the recessive model of rs12921862, the stratification analysis revealed that female cases had higher susceptibility than males for the AA genotype. Therefore, a further study with more samples needs to be conducted. The AA/AC and AC genotypes of rs12921862 in dominant and overdominant genetic models also present a correlation with a poor prognosis of DCM for survival analysis.
In general, according to our findings, these two SNPs of AXIN1 contribute to the susceptibility and prognosis of DCM. However, further studies are required to investigate the underlying mechanisms by which AXIN1 has an effect on DCM. In addition, the follow-up population needs to be expanded, since the present sample size is relatively small.
Conclusion
This study suggests that rs12921862 and rs1805105 of AXIN1 are associated with susceptibility to DCM. In addition, rs12921862 is also related to the prognosis of DCM in a Chinese Han population. Further studies and large samples are still indispensable to confirm our findings and to explore a more distinct mechanism of AXIN1 in DCM.
Footnotes
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81670346), the Applied Basic Research Programs of Science and Technology Commission Foundation of Sichuan Province (No. 2016SZ0013), the International Cooperation Project of Science and Technology Department of Sichuan Province (No. 2018HH0014), Science and Technology Major Project of Sichuan Province (No. 2017SZDZX0013), and Science and Technology Key R&D Project of Sichuan Province (2017FZ0077).
Disclosure Statement
No competing financial interests exist.