
Abstract
Cutis laxa represents a heterogeneous group of rare, inherited, or acquired connective tissue disorders with the common feature of loose and redundant skin with decreased elasticity. The skin of affected deer showed abnormal collagen fiber morphology. To identify the differentially expressed genes of the unusual localized skin laxity in sika deer, we performed transcriptome analysis in the affected and control sika deer. The transcriptome analysis showed 700 genes with significant differential expression in the affected skin as compared with normal skin. Pathway analysis revealed an enrichment of genes involved in tumor necrosis factor signaling, the extracellular matrix-receptor interaction, platelet activation, and Huntington's disease. A gene network was constructed, and the hub nodes such as PTGS2, THBS1, COL1A1, FOS, and NOS3 were found through PPI network analysis, which may contributed to the unusual localized skin laxity in sika deer. Abnormal expression patterns of genes during the development of the affected sika deer were successfully uncovered in the present study, which provides a reference for revealing the related mechanism underlying cutis laxa in sika deer and human beings.
Introduction
Sika deer (C
Cutis laxa (CL) is a heterogeneous group of rare, inherited, or acquired connective tissue disorders that share a common feature of loose and redundant skin with decreased elasticity. The typical clinical feature of CL in humans is the presence of the skin that can be easily pulled away from the underlying tissue which does not rapidly return to its original position after being released (Uitto and Pulkkinen,
There is less research regarding the organizational structure of the skin in deer. Previous studies evaluated the skin from eight different body parts of normal fallow deer (Dama dama), red deer (Cervus elaphus) and sika deer (Cervus nippon) using hematoxylin–eosin, Masson, or Orcein staining (Jenkinson, 1972). The dermis is divided into two layers: the upper layer is the papillary layer and the lower layer is the reticular layer, both of which join at the level of the sebaceous glands (Jenkinson, 1972). The rapid growth capacity of the skin and antlers of male deer is remarkable. Chunyi Li studied the skin in areas of antler growth; as the horns began to grow, it was noted that the first changes in the horns' skin were characterized by subcutaneous loose connective tissue compression, followed by corrugated skin stretching (Li and Suttie, 2000).
Current resources for the genomic sequencing of deer skin are scarce, especially for skin of affected sika deer. RNA-seq analyses have been carried out in various sika deer tissues, including those of the bone marrow (Yao et al., 2012c), antler tips (Yao et al., 2012b), and antlers (Zhao et al., 2013). The lack of sequence information for affected sika deer has limited the progress in gene discovery and characterization. Such an approach may aid in understanding the biological functioning of sika deer at the transcriptome level.
To understand the mechanisms that underlie the development of unusual localized skin laxity in sika deer, we first performed histology analysis and transcriptome sequencing. We compared gene expression patterns in unusual localized skin laxity in sika deer with those of control deer. The aim of our research was to clarify the foundational aspects of affected sika deer and to analyze the characteristics of the development of spontaneous disease in an animal model. In addition, we our study provides novel insights relevant to breeding and have profound impacts on deer farming.
Materials and Methods
Skin collection and tissue preparation
This study was carried out in strict accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health of China. The protocol was approved by the Animal Welfare Committee of the Institute of Special Animal and Plant of Sciences, China Academy of Agricultural Sciences (CAAS, Permit No.: 13–1006). All sedation and biopsy procedures were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
The animals were kept in a 60 m2 paddock and were fed kernel maize, 0.7 kg of bean pulp, and 2–3 kg of maize straw per head per day. Water was available ad libitum. Before examination, the deer were treated with 104 mg of Lumianbao, an anesthetic agent (xylazine; Qingdao Hanhe Animal & Plant Pharmaceutical Co., Ltd., China) using a homemade blowing needle with a 2 m long tube (Chen et al., 2015). One full-thickness (4 mm) punch biopsy was performed on the back of the neck of three male sika deer (4 years of age) of each affected and nonaffected (control) using skin biopsy punches (Electron Microscopy Sciences, Acuderm, Inc., Italy). We collected three skin samples from affected and nonaffected sika deer over a period of 3 years. The skins were cleaned with cold 1 × phosphate-buffered saline and then stored at −80°C.
Histology
The skin samples were fixed in 4% paraformaldehyde for 2 days and then dehydrated using a graded series of ethanol and embedded in paraffin. Eight millimeter sections were cut using a microtome (Leica, Germany). The collagen fibers were stained using Ponceau–acid fuchsin–aniline blue. The sections were washed with distilled water added with 10 drops of periodic acid solution for 5 min, rinsed, and incubated 3 min in the Harris hematoxylin solution. Sections were then rinsed in distilled water and immersed in the acid differentiation buffer for 3 s. They were rinsed again in distilled water and immersed in the reagent Ponceau–acid fuchsin (previously mixed in equal quantity) for 5 min. After rinsing in running tap water for 10 min, sections were put in the 2% aniline blue for 1 min, rinsed for 2–3 s in distilled water, and dehydrated through ascending alcohols, stopped for 1 min in the last absolute ethanol, and finally cleared in xylene and mounted.
Transcriptome sequencing, quality control, and read mapping
RNA was extracted from three skin samples taken from affected sika deer and three skin samples taken from the control sika deer using TRIzol reagent (Invitrogen). RNA sequencing was performed with 100 bp paired-end reads using an IlluminaHiSeq 2000 located at BGI-Shenzhen (Shenzhen, China). We removed reads that contained the adapter sequences, those containing >10% unidentified nucleotides and those in which >50% of the bases had a Phred quality score <5. The remaining clean reads were assessed for quality, error rate, and guanine plus cytosine content.
We used BLAST (Altschul et al., 1990) alignment of unigenes with NT (nucleotide database), NR (nonredundant database), COG (clusters of orthologous groups), KEGG, and Swiss-Prot to obtain the annotation; we also used Blast2GO (Conesa et al., 2005) with NR annotation to obtain the GO annotation, and InterProScan5 (Quevillon et al., 2005) to obtain the InterPro annotation. For the unigenes that were unannotated, we used ESTScan (Iseli et al., 1999) to predict the sequence coding for aminoacids in protein. We used MISA (Thiel et al., 2003) to find the simple sequence repeats (SSRs) in the unigenes, then designed primers for each SSR using Primer3 (Untergasser et al., 2012). We mapped all the clean reads to the unigenes using HISAT (Kim et al., 2015). We mapped the clean reads to the unigenes using Bowtie2 (Langmead and Salzberg, 2012), and then calculated the gene expression level with RSEM (Li and Dewey, 2011).
Analyses of differentially expressed genes
We used two programs, NOIseq and PossionDis, to compare the gene expression patterns of the affected sika deer and the control sika deer. The NOIseq program is based on a noisy distribution model and was performed as described by Tarazona et al. (2011) with a fold change ≥2.0 and a probability ≥0.8. The PossionDis program is based on the Poisson distribution and was performed as described by Audic and Claverie (1997) with a fold change ≥2.0 and an false discovery rate (FDR) ≤0.001 (). To decrease the rate of false positives, only those genes that were identified by both programs were considered to have a significant difference in expression levels.
Based on the GO and KEGG annotation results, we classified the differential expression genes (DEGs) according to their official classification, and we also performed GO and pathway functional enrichment using phyper, a function within the R software (Cheng et al., 2017). We calculated the FDR for each p-value, and the terms for which FDR was not >0.01 were defined as significantly enriched.
Protein and protein interaction network construction
Protein and protein interaction (PPI) analysis was performed on DEGs using the STRING online tool. 1 Medium confidence (combined score) >0.4 was selected as the threshold. Cytoscape_3.4.0 was used for the construction of a PPI work.
Quantitative real-time PCR validation of mRNA
We performed reverse transcription quantitative polymerase chain reaction (PCR) to verify the expression of the genes of interest. All primers were designed based on identical sequences of the relevant genes from sika deer (Supplementary Table S1). The prepared cDNA was amplified with a FastStart Universal SYBR Green Master (Rox) (Roche Diagnostics, Germany) and gene-specific primers were used with a final reaction mixture of 25 μL. Amplification and quantification were performed with an Exicycler 96 Quantitative PCR Analyzer (Applied Biosystems) under the following cycling conditions: preincubation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. Melt curves for each gene were analyzed to ensure the amplification of single PCR products. In addition, agarose gel (1.5%) electrophoresis was performed to determine the length of the amplified PCR products. GAPDH was used for normalization. Each experiment was independently repeated three to four times, and fold changes in gene expression were analyzed by the △△Ct method.
Results
Histological features of affected skin
The histological examination of the skin sections from the affected deer (Fig. 1A) (Chen et al., 2018) stained with Ponceau–acid fuchsin–aniline blue revealed collagen fibers with an abnormal morphology, in which the reticular dermis had a marked grossly granular, fragmented, and clumped appearance. In addition, markedly reduced quantities of reticular collagen fibers were observed. The staining of skin biopsy sections from the control deer revealed a normal quantity of reticular elastin. Collagen fibers were observed as tiny, closely arranged fine fibers that formed scattered bundles in the reticular dermis of normal skin, with a deep reticular layer of thick, mainly parallel fibers (Fig. 1B).

Genes with differential expression between the two skin morphologies
We generated the raw and clean reads in affected deer and the control sika deer samples. The quality metrics for the assembly are given in Supplementary Table S2. The quality of the resulting reads, after trimming and filtering, is given in Supplementary Table S3. We cluster transcripts into unigenes, and the quality metrics for the clustering are given in Supplementary Table S4.
After assembly, we performed functional annotation of the unigenes using seven functional databases (NR, NT, GO, COG, KEGG, Swiss-Prot, and InterPro); the annotation summary is given in Supplementary Table S5. For NR annotation, the distribution of the annotated species was statistically analyzed, as given in Supplementary Figure S1. The unigenes shared between the GO, and KEGG annotations and their functional distribution were statistically analyzed, as given in Supplementary Figures S2 and S3, respectively.
The gene expression analyses showed significant differences in gene expression between the affected sika deer and the control sika deer. We identified 700 genes with statistically significant differences in expression between the two skin morphologies, including 228 upregulated genes and 472 downregulated genes in the affected sika deer relative to the control sika deer (Fig. 2A).
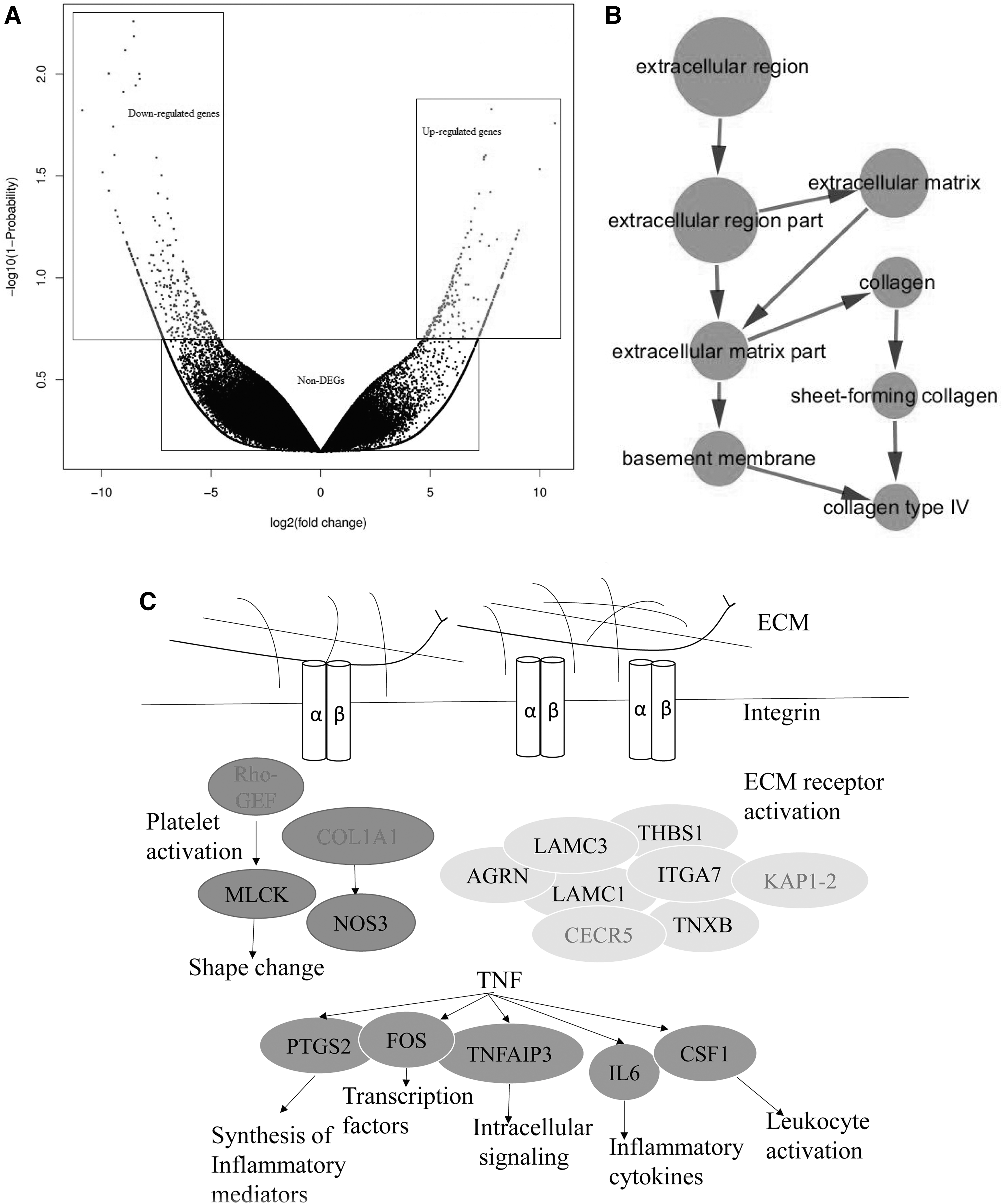
GO and KEGG pathways analysis of DEGs from different skin morphologies
In the GO analysis, the functional categories of these genes were clustered mostly within the top 50 GO terms. The top enriched terms in the cellular component categories were concentrated in collagen type IV (Fig. 2B). We found that the DEGs were mainly associated with cell, cell part, cellular process, organelle, binding, and metabolic process. The KEGG enrichment of the differentially expressed genes were related to the tumor necrosis factor (TNF) signaling pathway (ko04668; Supplementary Fig. S4), rheumatoid arthritis (ko05323), regulation of lipolysis in adipocytes (ko04923), and the extracellular matrix (ECM)-receptor interaction (ko04512; Supplementary Fig. S5) (Table 1, Fig. 2C).
Differential Expression of Selected Genes Grouped by Pathway Between the Affected Sika Deer and the Control Deer
The regulatory networks involved in unusual localized skin laxity in sika deer
We used STRING to obtain PPI relationships (Supplementary Fig. S6). The connectivity degree of PTGS2 (prostaglandin-endoperoxide synthase 2), THBS1 (thrombospondin 1), COL1A1 (collagen, type I, alpha 1), FOS (FBJ murine osteosarcoma viral oncogene homolog), and NOS3 (nitric oxide synthase 3) were ≥9 and were identified as hub genes. They might play important roles in unusual localized skin laxity in sika deer (Fig. 3).

Connectivity degrees of hub genes related to unusual localized skin laxity in sika deer analyzed the protein and protein interaction network.
Validation of RNA-seq results by quantitative real time polymerase chain reaction
In addition, the changes in the expression of seven randomly selected DEGs showed positive correlations with the quantitative real time polymerase chain reaction (qPCR) results, and their expression was determined using RNA-seq (Fig. 4). There were significant differences in the expression of seven genes in the affected sika deer relative to the control deer, including PTGS2 and FOS, which are related to the TNF signaling pathway, THBS1, COL1A1, and TNXB, which are related to ECM-receptor interaction, NOS3, which is involved in the platelet activation signaling pathway, and λ-IgLC, which is involved in the Huntington's disease signaling pathway (Table 1).
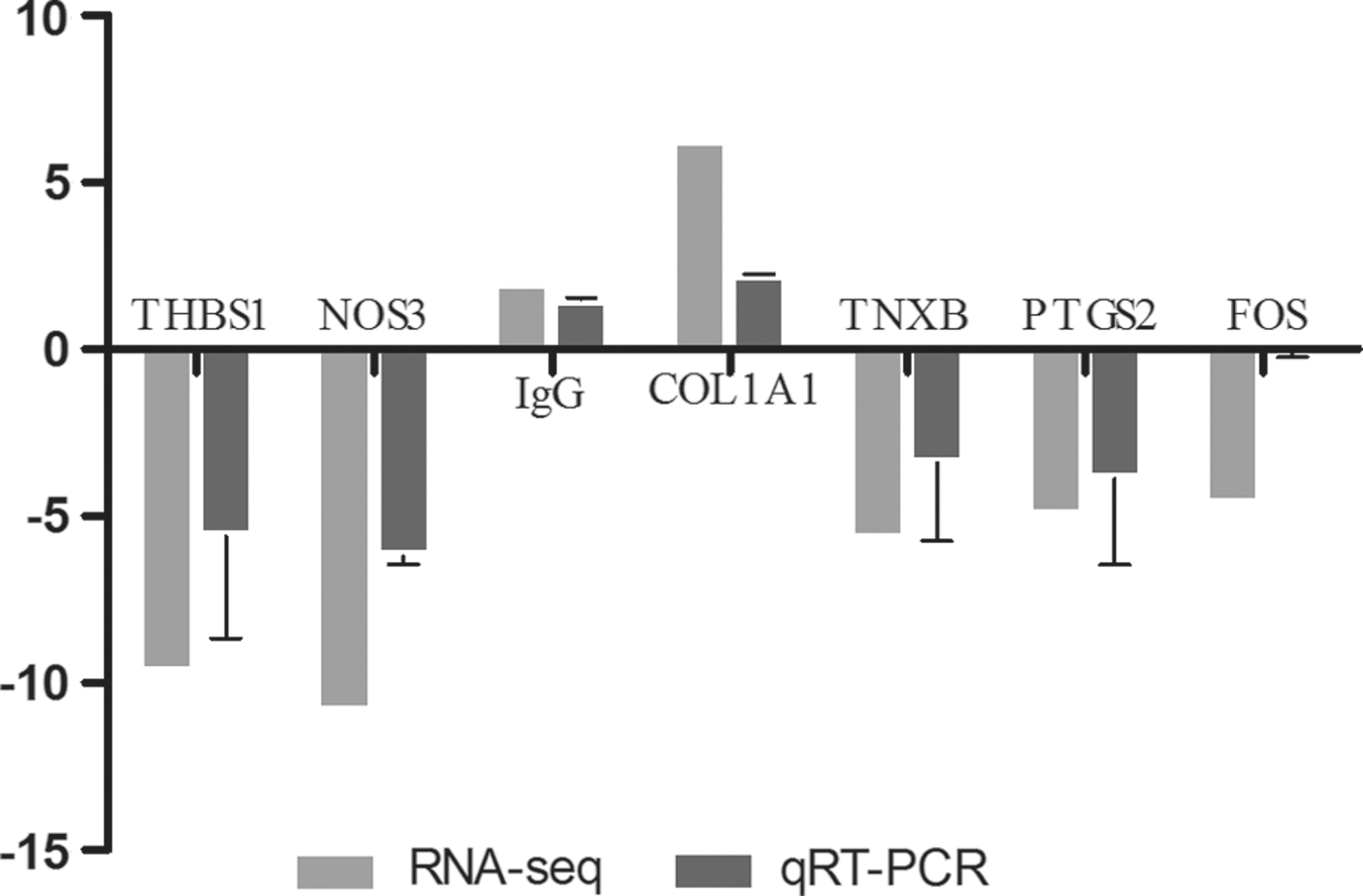
Validation of RNA-Seq data using qRT-PCR. All results were compared with the control group and presented as mean ± SEM for three different samples. qRT-PCR, quantitative real time polymerase chain reaction.
Discussion
This is the first report containing transcriptome data obtained from sika deer skin. Compared with other deer transcriptomes within Cervidae (Yao et al., 2012a), the de novo assembly and annotation of sika deer transcriptome were superior. Although sika deer skin is similar to that of other mammals (Jenkinson, 1972), there may be differences between CL in humans and sika deer. All deer in our study were males, so it is possible that skin laxity inheritance could be linked to the X chromosome, as in occipital horn syndrome. The affected deer in this study did not have any other symptoms, with the exception of greater weight of the antler velvet compared with that of normal deer (Chen et al., 2018). We found abnormal collagen fibers in affected sika deer that were similar to fibers observed in human patients with CL. The collagen fibers had a marked, grossly granular and a microscopically fragmented and clumped appearance. In addition, Jenkinson (1972) found that the fallow deer (Dama dama), red deer (Cervus elaphus), and sika deer (Cervus nippon) have the same skin structure as other mammals. There are other animal models of CL that contribute to the current understanding of CL and provide an animal model of ARCL1C (Bultmann-Mellin et al., 2015). Given such a large number of contributing genes, we applied a population approach to infer the possible functional consequences by observing the enrichment of the GO and KEGG pathway annotation terms in the corresponding set.
The KEGG functional analysis indicates that the affected deer is associated with immune diseases and the immune system. Among the pathways, the greatest number of assembled unigenes was found to be involved in the signaling transduction pathway that has recently been shown to play a central role in hair follicle and skin development (Rishikaysh et al., 2014). Of note, the TNF signaling pathway was the most enriched pathway in the affected sika deer. TNF is a critical cytokine that can influence a wide range of intracellular signaling pathways, including those involved in apoptosis and cell survival as well as inflammation and immunity.
Our transcriptome analysis identified significant differences in the expression of a number of unusual localized skin laxity-related genes, including THBS1, COL1A1, TNXB, NOS3, FOS, PTGS2, LAMB1, and λ-IgLC, between the affected and control sika deer. Several other genes, such as FBLN5 (Elahi et al., 2006), FBLN4 (Hucthagowder et al., 2006), PYCR1 (Reversade et al., 2009), LTBP-4 (Bultmann-Mellin et al., 2015), ATP6V0A2 (Kornak et al., 2008), ALDH18A1 (Wolthuis et al., 2014), EFEMP2 (Papke et al., 2015), and ELN (Callewaert et al., 2011), have been identified in previous studies to be related to CL in humans; however, these genes were not found to be differentially expressed in this study. This inconsistency indicates that the molecular programs, which are complex processes involving numerous signaling pathways, genes, reciprocal signaling interactions and flexible regulatory networks, that contribute to unusual localized skin laxity are highly species specific.
Among the genes that we identified, COL1A1 and λ-IgLC, which are involved in the ECM-receptor interaction pathway and the Huntington's disease pathway, respectively, were upregulated. The dominant variants of COL1A1 and COL1A2 are known to cause EDS phenotypes (Malfait and De Paepe, 2014), and it has also been found that COL1A1 causes EDS in a recessive manner (with the parents in both families being completely clinically normal) through exome sequencing, which was performed by a clinical laboratory using probands from two families and revealed the same novel homozygous missense variant in COL1A1 (c.2050G>A,
In this study, we identified five DEGs as hub genes that are likely more important than other genes because of their key positions in the network. Candidate genes such as THBS1, TNXB, NOS3, PTGS2, FOS, and LAMB1 have been shown to be downregulated in the affected sika deer. Conversely, the levels of FOS gene expression did not differ from those observed in normal fibroblasts (Hatamochi et al., 1996). THBS1 expression is also upregulated and is involved in antler growth (60 days vs. 90 days) (Zhao et al., 2013). This finding is inconsistent with our results. Previously, it was found that one of the roles of THBS1 was the activation of transforming growth factor β (TGFβ) during wound healing, and that THBS1 is involved in the pathogenesis of fibrotic processes in the kidney and heart in patients with diabetes (Belmadani et al., 2007). During skin development, increased expression of thrombospondin and TGFβ activity was observed in fibrotic skin disorders, such as keloids (Chipev et al., 2000) and scleroderma (Mimura et al., 2005). We can speculate that decreases in the expression of THBS1 are presumably the result of its participation in the development of CL in sika deer. In regard to TNXB in deer, previous research found that joint hypermobility and fibrillary collagen perturbations because of mutations in TNXB will cause CL (Tokhmafshan et al., 2016). Other studies on CL have indicated that mutations in fibulin-4 cause CL and thoracic aortic aneurysms. The expression of PTGS2 is upregulated in the descending aorta of fibulin-4 knockout mice (Fbln4−/− AA) compared with Fbln4+/+ AA and Fbln4−/−, mice, suggesting that ERK1/2 activation, degradation of collagen type 1, and increased inflammation contribute to the pathogenesis of aneurysms (Jungsil Kim et al., 2015). LAMB1 has been suspected to be involved in CL, and antihuman laminin antiserum demonstrated that this protein is absent from the skin (Fischer-Zirnsak et al., 2015). In sika deer, THBS1, TNXB, NOS3, PTGS2, FOS, COL1A1, LAMB1, and λ-IgLC were identified and then used to explore the molecular genetic basis of CL in animals.
Conclusions
In summary, these results demonstrate for the second time the unusual localized skin laxity in sika deer. Novel expression patterns of genes during the skin development of the affected sika deer were successfully verified with DEGs and quantitative PCR. Our findings provide new insights relevant to CL and have profound impacts on deer farming. Further investigation on functional of DEGs in affected deer will be necessary to declare its molecular mechanisms in vivo and in vitro.
Footnotes
Acknowledgments
The authors thank Yalin Cheng for helpful discussion and comments on the earlier versions of this article. The authors also thank Hailong Xue, Xuezhe Cui, and Min Chen for assisting in the sample collection. The authors sincerely thank the editor and reviewers for their valuable comments. This work was supported by the Jilin Department of Science and Technology of China (20170623036TC) and Jilin Department of Science and Technology of China (20180201043NY).
Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.