
Abstract
Interstitial lung disease (ILD) is the main reason of death in patients with systemic sclerosis (SSc). The potential microRNA (miRNA)–messenger RNA (mRNA) interaction networks of SSc-ILD from a systematic biological perspective are unclear. To characterize differentially expressed miRNAs (DE-miRNAs) and differentially expressed genes (DEGs) likely related to SSc-ILD, we downloaded the miRNA microarray dataset (GSE81923) and mRNA datasets (GSE76808 and GSE81292) from the Gene Expression Omnibus database. Comprehensive bioinformatic analyses were conducted to predict target genes for DE-miRNAs and generate an miRNA-hub gene network with SSc-ILD. In total, 26 DE-miRNAs were detected in SSc-ILD, among which 2 were upregulated and 24 were downregulated. Additionally, 178 common DEGs (55 upregulated and 123 downregulated) were identified. miRNAs were primarily enriched in pathways involving inflammation and regulation of fibroblasts. The hub genes identified were MMP7, IER2, HBEGF, CCL4, NFKBIA, JUNB, LIF, SERPINE1, FOSL1, and NAMPT. We discovered the miRNA-mediated regulatory network in SSc-ILD using an integrated bioinformatic analysis. The findings provide novel insight and expand our comprehension of the molecular mechanisms participating in the pathogenesis of SSc-ILD, along with identification of new potential diagnostic biomarkers.
Introduction
Systemic sclerosis (SSc) is an uncommon disease of connective tissues that is specified by excessive collagen deposition, microvascular involvement, and autoimmunity (Servettaz et al., 2006). Pulmonary involvement, particularly interstitial lung disease (ILD), results in high mortality rates in patients with SSc (Elhai et al., 2012). Approximately 79% of SSc patients are found to have ILD during autopsy (D'Angelo et al., 1969). One multicenter observational study of over 5000 patients with SSc showed that ILD accounted for more than one-third of SSc-related deaths (Tyndall et al., 2010). The prevalence of SSc-ILD is high, with more than 15% of patients progressing to severe lung fibrosis. The mechanism and etiology in SSc-ILD formation and progression have been investigated extensively, yet causes of SSc-ILD remain unclear. Distinguishing between individuals with severe disease and patients with slow or stable disease makes it challenging for physicians to identify appropriate therapies (Roth et al., 2011).
microRNAs (miRNAs), known as a class of short noncoding RNAs, have the capability of negatively regulating the expression of genes through interactions with the 3′-untranslated region of the targeted messenger RNA (mRNA) (Wang et al., 2018). miRNAs are recognized for their important roles in numerous cellular processes, such as cellular apoptosis, proliferation, and differentiation (Baumjohann and Ansel, 2013; Wang et al., 2018). To date, miRNAs and their association with pulmonary fibrosis have become a central topic for many biomedical researchers around the globe. miRNA and mRNA show excellent stability and reliability in the blood, which increases their potential use as novel markers for detection and monitoring of SSc-ILD in the clinic (Young et al., 2017). In the past decade, technologies classified as “-omics” have been widely utilized to characterize alternations of gene expression and identify potential biomarkers at the molecular level with detailed insights into the disease (Kim et al., 2006; Milano et al., 2008). SSc-ILD is thought to be associated with multiple putative miRNAs, including miR-29a (Khalil et al., 2015), miR-155 (Christmann et al., 2016), miR-21, miR-182 (Kouri et al., 2015), and miR-92a (Bagnato et al., 2017). The different reports on modified miRNA expression were primarily caused by sample heterogeneity, different sequencing platforms, and distinct analytical approaches. However, the relationship between SSc-ILD and miRNA-mRNA interaction networks remains unclear from a systematic biological perspective.
Microarray datasets were retrieved from the Gene Expression Omnibus (GEO) database to investigate correlations between differentially expressed genes (DEGs) and differentially expressed miRNAs (DE-miRNAs) by bioinformatic analyses. This allowed for characterization of DE-miRNAs in the miRNA expression profiles, along with DEGs in the mRNA expression profiles of patients with SSc-ILD. The DE-miRNA and DEG data were combined for the gene ontology (GO) and pathway enrichment analysis along with functional module analyses of protein–protein interaction (PPI) networks. The interaction network between miRNA and mRNA associated with SSc-ILD provides the fundamentals for expanding our knowledge of biological mechanisms underlying the pathogenesis of SSc-ILD and for identification of diagnostic and therapeutic targets of SSc-ILD.
Materials and Methods
Data collection and inclusion criteria
The National Center for Biotechnology Information (NCBI) GEO database* is a public genomics data repository that contains gene expression datasets and original series and platform records. Microarray data from Idiopathic pulmonary fibrosis (IPF)-relevant miRNA and mRNA expression profiles were obtained and extracted from the GEO database. Keywords, including “Systemic sclerosis,” “interstitial lung disease,” “Homo sapiens,” and “expression profiling by array,” were used to search for publicly available studies, from which 11 GSE studies were retrieved after a systematic review and screened with the inclusion criteria as follows: (1) samples diagnosed with SSc-ILD tissues and normal tissue samples and (2) gene expression profiling of mRNA or miRNA. Finally, the miRNA microarray dataset, GSE81293 (Christmann et al., 2016), and the mRNA microarray datasets, GSE76808 and GSE81292 (Christmann et al., 2014), were used for further analysis. Baseline characteristics and demographic information of patients with SSc-related ILD and healthy controls were previously described by Christmann et al. (2014, 2016).
Microarray data information
The microarray dataset, GSE81293, was from the GPL16384 platform ([miRNA-3] Affymetrix Multispecies miRNA-3 Array) with lung biopsy tissues from 20 matched SSc-ILD and normal lung samples. The dataset, GSE76808, was from the GPL571 platform ([HG-U133A_2] Affymetrix Human Genome U133A 2.0 Array) with samples from seven SSc-ILD patients and four healthy persons. The GSE81292 dataset was from the GPL18991 platform ([HG-U133A_2] Affymetrix Human Genome U133A 2.0 Array), containing 15 lung biopsy samples of SSc-ILD and 5 healthy persons.
Data processing and characterization of DE-miRNAs and DEGs
Data were preprocessed for background adjustment, normalization, and summarization in R language (version 3.5.0). Probe levels were calculated and converted into gene expression levels according to the annotation files in the GEO database. DE-miRNAs and DEGs between the SSc-ILD and normal controls were analyzed with the limma package (version 3.34.8) in the R language (Smyth, 2004). p-Values were adjusted by the Benjamini–Hochberg false discovery rate method, and adjusted p-value (adj. p) <0.05 and |log fold change (FC)| >1 were used as the cutoff values. The heat map of DEGs was generated using the pheatmap function of the R/Bioconductor package (Reimers and Carey, 2006). A volcano plot of DE-miRNAs and DEGs was drawn using the ggplot2 package (version 3.1.0).
Functional and pathway enrichment analysis
GO for DEGs was performed to assign the genes and their products into biological processes (BPs), molecular functions (MFs), and cellular components (CCs). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was utilized with the Database for Annotation, Visualization, and Integrated Discovery (DAVID) ** ; to identify functional and metabolic pathways (Huang Da et al., 2009). A bubble chart was generated using the ggplot2 package (version 3.1.0). GO and pathway enrichment analyses of DE-miRNAs were applied by DIANA miRPath (Vlachos et al., 2012), and any p-value <0.05 was rated to be statistically significant.
PPI network and detection of hub genes
The Search Tool for the Retrieval of Interacting Genes (STRING) ** is an online database for discovering gene interactions, which includes both physical and functional associations (Szklarczyk et al., 2011). STRING was used to evaluate interactive associations among DEGs with a confidence score of >0.4 as the cutoff criterion. Cytoscape software (version 3.5.1) (Shannon et al., 2003) was applied to construct the PPI network. The hub genes in the network were detected to characterize key elements in SSc-ILD, using cytoHubba (version 0.1) in Cytoscape (Chin et al., 2014).
Prediction of miRNA targets
To understand the associations between miRNAs and gene expression, the genes targeted by DEGs were predicted from GSE81293 using TargetScan † (Lewis et al., 2005), miRanda (John et al., 2004), and miRDB ‡ tools (Wong and Wang, 2015). Genes from three prediction methods were considered as the candidate targets. The mRNA-miRNA interactions were generated by Cytoscape to further characterize correlations between miRNAs and potential target genes (Kohl et al., 2011).
Results
Baseline characteristics of patients with SSc-related ILD
All patients with SSc-related ILD from the mRNA microarray datasets, GSE76808 and GSE81292 (Christmann et al., 2014), were female. More than half (52%) of the patients had Diffuse cutaneous SSc (dcSSc) and 48% were positive for the anti-Scl-70 (antitopoisomerase I) antibody. Most patients with SSc-ILD developed progressive disease in the miRNA microarray dataset GSE81293 (Christmann et al., 2016) and showed an average baseline score of 6.88 ± 4.47 compared with 12.76 ± 6.59 after treatment. The detailed characteristics and demographic information of patients with SSc-related ILD and healthy controls were previously described by Christmann et al. (2014, 2016).
Detection of DEGs in two mRNA microarray datasets
As shown in Table 1, a total of 29 SSc-ILD and 9 matched normal lung tissues were analyzed, from which 356 and 413 DEGs were identified from datasets GSE76808 and GSE81292, respectively, using adj. p < 0.05 and |log FC| >1 as the cutoff. Among them, 139 DEGs were upregulated and 217 were downregulated in GSE76808 (Fig. 1a, b), while 165 were upregulated and 248 were downregulated in GSE81292 (Fig. 1c, d). As shown in Table 2, a total of 178 DEGs were identified by the integrated analysis, including 55 upregulated DEGs and 123 downregulated DEGs between the SSc-ILD and healthy controls.

Identification of DEGs between SSc-ILD and healthy controls.
Dataset Details of the microRNA and Gene Expression Profiles Related to Patients with Systemic Sclerosis–Interstitial Lung Disease
GEO, Gene Expression Omnibus.
One Hundred Seventy-Eight Differentially Expressed Genes Identified in Patients with Systemic Sclerosis–Interstitial Lung Disease
DEG, differentially expressed gene.
GO and pathway enrichment analysis of DEGs
As shown in Figure 2a, 178 DEGs were annotated and mainly classified into BPs, MFs, and CCs. In the BP category, DEGs were significantly enriched in the organic cyclic compound, regulation of BPs, regulation of cell processes, and cell proliferation. In the CC group, GO terms were higher in the extracellular space, extracellular region, extracellular matrix (ECM), cell surface, and cytoplasm. In the MF category, DEGs were enriched in cytokine activity, transcriptional activator activity, receptor binding, cytokine receptor binding, chemokine activity, core promoter proximal region sequence-specific DNA binding, and RNA polymerase II core promoter proximal region sequence-specific DNA binding. Next, 178 DEGs were enriched in 34 signal pathways, including the tumor necrosis factor (TNF) signaling pathway, cytokine–cytokine receptor interaction, NF-kappa B signaling pathway, NOD-like receptor signaling pathway, Toll-like receptor (TLR) signaling pathway, rheumatoid arthritis, and MAPK signaling pathway. The top 20 pathways are shown in Figure 2b.
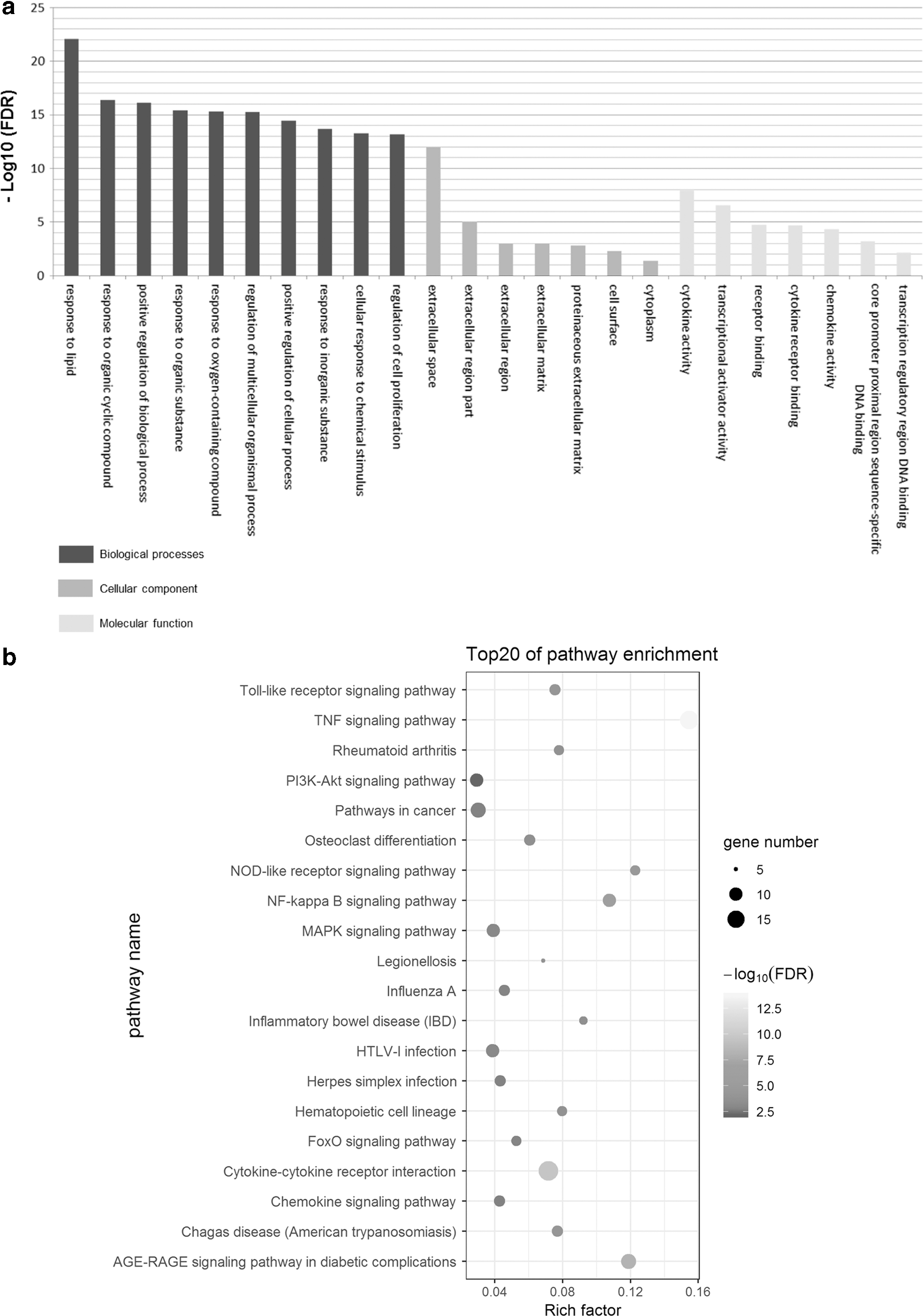
GO term and KEGG pathway enrichment analysis of 178 DEGs.
Construction of the PPI network and hub gene analysis
The PPI network of 178 DEGs, which contained 177 nodes and 522 edges, was built using the Cytoscape software based on the STRING database (Fig. 3a). The hub genes were characterized by the Cytoscape software (Fig. 3b). The 10 top hub nodes with the highest degrees of connectivity were matrix metallopeptidase 7 (MMP7), immediate early response 2 (IER2), heparin-binding EGF-like growth factor (HBEGF), CC motif chemokine ligand 4 (CCL4), NFKB inhibitor α (NFKBIA), JunB proto-oncogene (JUNB), leukemia inhibitory factor (LIF), interleukin 6 family cytokine, serpin family E member 1 (SERPINE1), FOS-like 1, AP-1 transcription factor subunit (FOSL1), and nicotinamide phosphoribosyltransferase (NAMPT).
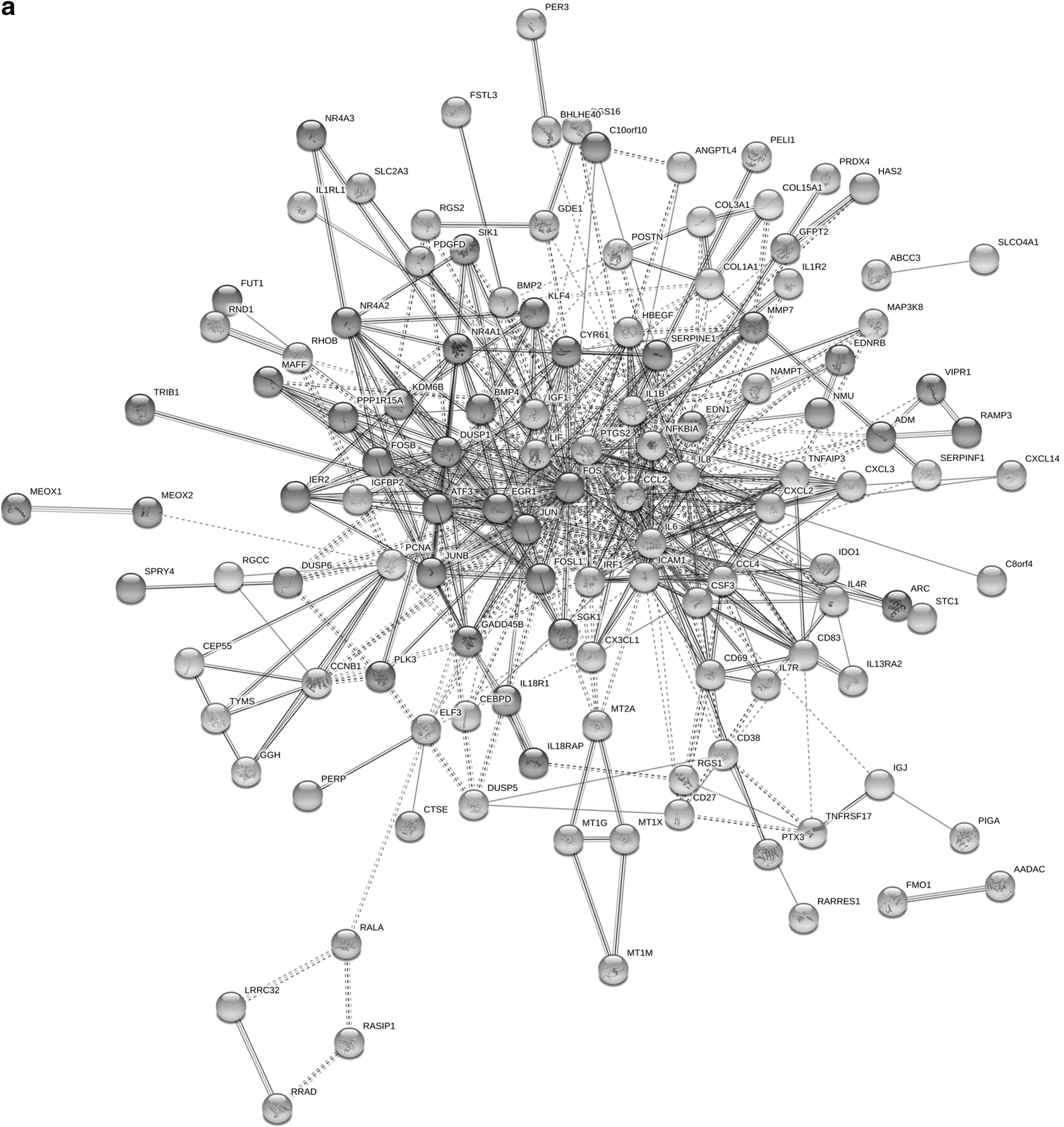
PPI network and hub genes of potential DEGs.
DE-miRNA pathway enrichment analysis
A total of 26 DE-miRNAs were identified from GSE81293 expression profiling datasets by comparing the SSc-ILD samples with healthy controls. Among them, 2 miRNAs (hsa-let7e and hsa-miR-455) were significantly upregulated, while the other 24 miRNAs were downregulated in the SSc-ILD samples (Table 3). The top 10 dysregulated miRNAs were hsa-miR-4459, hsa-miR-4507, hsa-miR-4417, hsa-miR-3687, hsa-miR-1246, hsa-miR-4668, hsa-miR-4530, hsa-miR-4299, hsa-miR-4516, and hsa-miR-663b. The KEGG analysis showed that 26 DE-miRNAs were enriched in 26 signaling pathways (p < 0.05), of which 20 most significantly enriched pathways, as determined by p-values, are shown in Figure 4a. GO term analysis demonstrated that DE-miRNAs were enriched in 175 functional GO terms, with the 20 most common shown in Figure 4b. A heat map of selected pathways and GO categories of the union genes are shown in Figure 4.
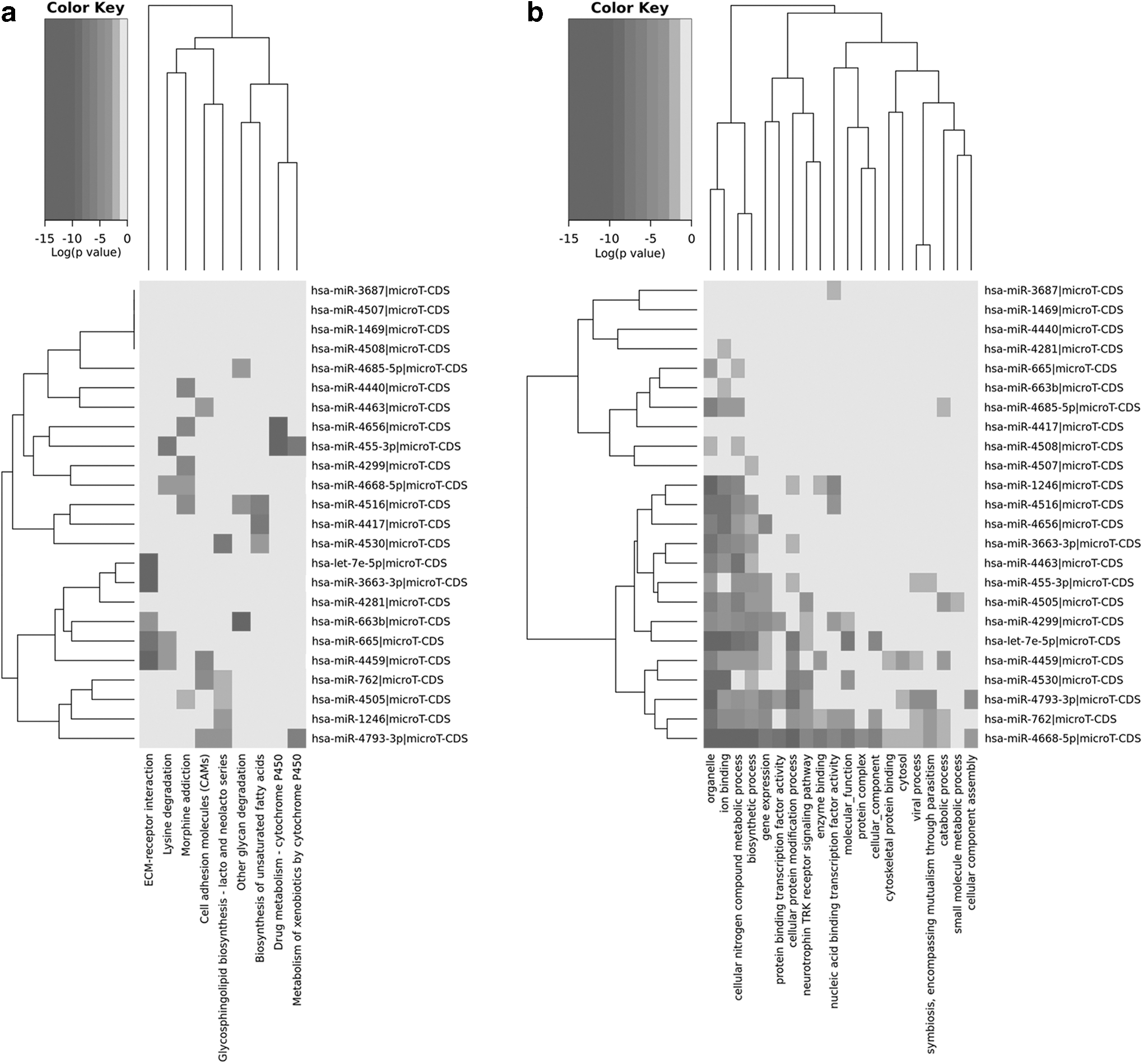
Heat map of DE-miRNAs in the GSE81293 dataset.
Twenty-Six microRNAs Significantly Expressed in Systemic Sclerosis–Interstitial Lung Disease from the GSE81293 Dataset
FC, fold change.
Correlation between DE-miRNAs and DEGs associated with SSc-ILD
The candidate target genes were predicted from the 26 dysregulated miRNAs using three miRNA-target interaction tools, including miRanda/mirSVR, TargetScan, and miRDB. Intersections between the candidate target genes and 178 common DEGs from three datasets were determined. As shown in Table 4, 14 miRNAs and their target genes commonly existed in DEGs of the two mRNA datasets. In total, six DEGs of the hub genes that were commonly predicted from two mRNA datasets were regulated by dysregulated miRNA (Table 5). The C-X3-C motif chemokine ligand 1 (CX3CL1) was predicted as the target of three miRNAs, and Fos-related antigen 1 (FOSL1) and FOSB (known as G0/G1 switch regulatory protein 3, G0S3) as targets of two miRNAs. Cytoscape was used to generate the miRNA-gene network for further elucidation of correlations between miRNAs and potential target genes (Fig. 5).
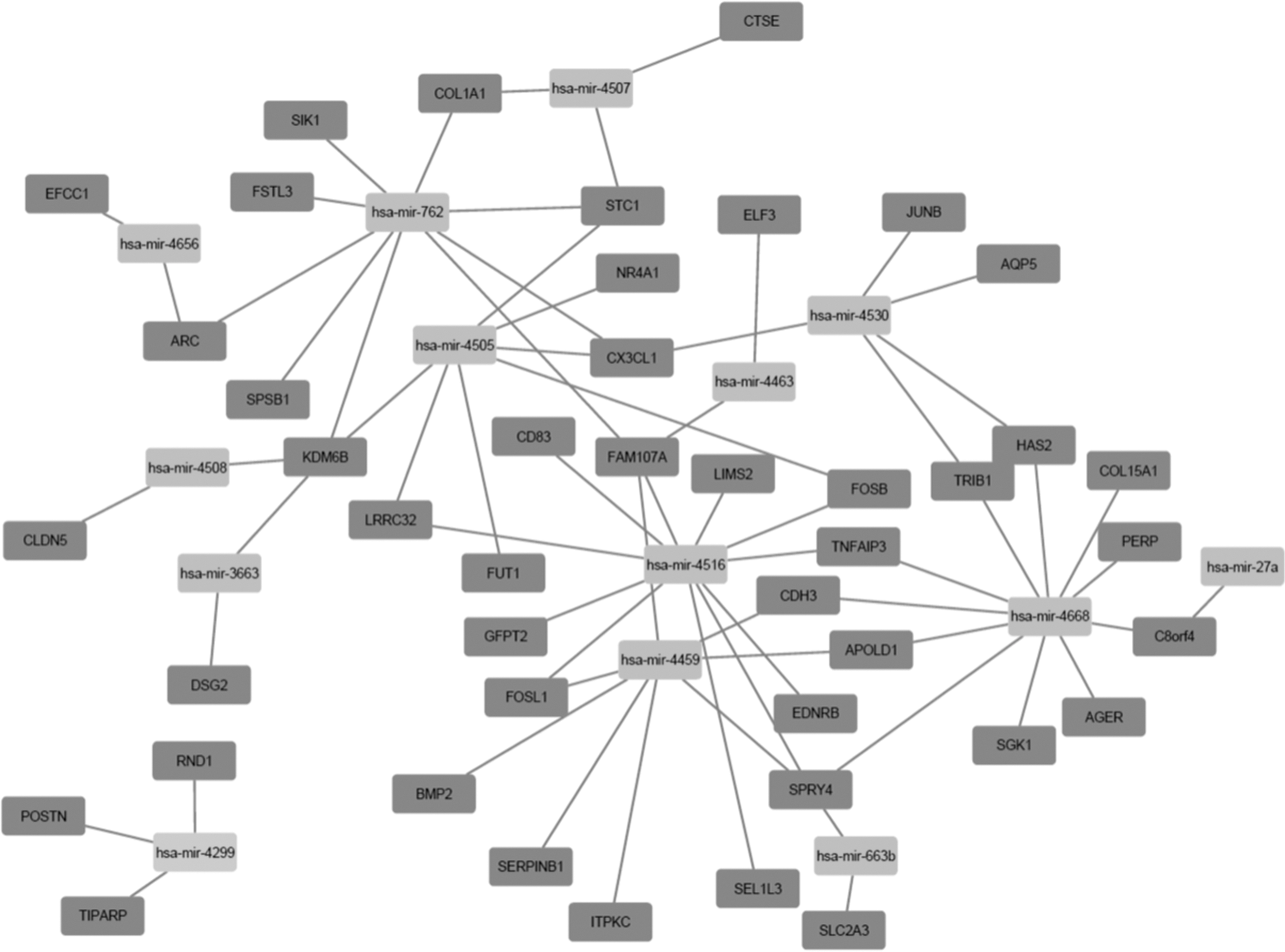
The regulation network of DE-miRNAs and DEGs in SSc-ILD.
Fourteen microRNAs and Their Target Genes Found in Differentially Expressed Genes of the Two Common Messenger RNA Datasets
Dysfunctional microRNAs Regulated Six Differentially Expressed Genes in the GSE81293 Dataset
Discussion
ILD is a common presentation of SSc with high mortality rates, yet the pathogenesis and optimal treatment of SSc-ILD remain unclear. Recently, several miRNAs, including the miR-29 family, miR-30 family, and let-7d (Tanaka et al., 2013; Bagnato et al., 2017), were found to be involved in the pathogenesis of many human diseases, such as cancer and inflammatory diseases (Davidson-Moncada et al., 2010). In addition, these miRNAs were shown to modulate several fibrotic-related genes with multifunctional roles in SSc fibrosis (Angiolilli et al., 2018). The overexpression of miR-29a in SSc fibroblasts reduces the expression of antiapoptotic proteins, including BCL-2, increasing the susceptibility to cell death (Jafarinejad-Farsangi et al., 2015). The inhibition of profibrotic miRNA miR-21, which is commonly overexpressed in SSc, results in increased apoptosis in SSc fibroblasts (Jafarinejad-Farsangi et al., 2016). In addition, miR-30-induced therapies were demonstrated by Tanaka et al. (2013) in patients with IPF and SSc. In the present study, 26 DE-miRNAs (2 upregulated and 24 downregulated miRNAs) were identified from the miRNA expression microarray dataset with SSc-ILD and control lung tissue samples (Table 3) and were most significantly enriched in the signaling pathways associated with inflammation and fibroblast regulation, such as ECM-receptor interaction, transforming growth factor-beta (TGF-β) signaling pathway, Wnt signaling pathway, and cell adherence junctions (Fig. 4).
Any upregulated miRNA typically leads to declined production of the target protein, while downregulation often leads to an excess in production of target proteins (Makino and Jinnin, 2016). Previous studies revealed the effects of miRNAs on fibrogenic activity by targeting TGF-β signaling events in lung epithelial cells and fibroblasts and regulating target genes in lung inflammation and lung fibrosis processes (Lepparanta et al., 2012; Christmann et al., 2014). ECM-receptor interactions are closely related to IPF (Hoffmann et al., 2014). In addition, let-7 regulated epithelial–mesenchymal transition and TGF-β to participate in pulmonary fibrosis (Pandit et al., 2010). MiR-92a decreased TGF-β1-induced Wnt1 inducible signaling pathway protein 1 (WISP1) expression in lung fibroblasts ex vivo (Lappi-Blanco et al., 2013; Berschneider et al., 2014). As cell adhesions and biological adhesions are critical processes for the formation of pulmonary fibrosis (Lappi-Blanco et al., 2013), we hypothesized that DE-miRNAs identified in this study might be associated with progression of SSc-ILD. Future studies are required to functionally and clinically validate DE-miRNAs in progression of SSc-ILD.
Microarray and high-throughput screening technologies have been widely applied to predict new target genes. However, previous studies have focused on a single genetic event or individual cohorts with limited sample sizes. In the present study, miRNA expression microarray data from two cohorts were collected, from which a total of 178 DEGs (55 upregulated and 123 downregulated) were detected. Signaling pathway analysis revealed associations of the cytokine–cytokine receptor interaction and NF-kappa B and TLR signaling pathways with progression of ILD in SSc patients. Innate immune signaling by TLRs has been generally accepted as a crucial factor that drives the persistent fibrotic response in SSc (Bhattacharyya and Varga, 2015). The KEGG analysis and preliminary bioinformatic analysis confirmed the cytokine–cytokine receptor interaction signaling pathway as a critical inflammatory regulator associated with chronically inflamed fibrosis. In addition, many hub genes were characterized by the PPI network construction for the DEGs, with the top 10 being MMP7, IER2, HBEGF, CCL4, NFKBIA, JUNB, LIF, SERPINE1, FOSL1, and NAMPT. Future studies are needed to validate these important hub genes as potential prognostic markers and therapeutic targets for SSc-ILD.
We reported possible joint action of DE-miRNAs and DEGs in SSc-ILD. In the present study, miRNA-mRNA interactions were analyzed using miRNA and mRNA expression profiles to gain additional insight into the essential roles of miRNAs in regulating progression of lung fibrosis in SSc. In total, 14 miRNAs showed regulatory effects on the identified DEGs that were overlapped between the 2 mRNA datasets and 10 DE-miRNAs and 6 DEGs involved in this miRNA-target relationship (Tables 4 and 5; Fig. 5), suggesting possible coregulation of DE-miRNAs and DEGs in SSc-ILD. Among them, CX3CL1 was predicted as the target of three miRNAs, and FOSL1 and FOSB as the target of two miRNAs. CX3CL1 is generated by type 2 pneumocytes and airway epithelium (Marasini et al., 2005) and forms the biological axis (CX3CR1/CX3CL1) with CX3CR1 to recruit plasma cells to the interstitium for generation of autoantibodies that can damage the parenchyma of SSc-ILD lungs. Recently, a large cohort study demonstrated associations of CX3XL1 with antitopoisomerase I antibody and lung fibrosis, and eventually with ILD progression, using multivariable regression analysis (Hoffmann-Vold et al., 2018). Both FOSL1 and FOSB genes encode leucine zipper proteins that are members of the FOS gene family and enable dimerization of proteins in the JUN family, including JUNB, to regulate cell proliferation, differentiation, and transformation. JUN and FOS were positively expressed in fibrous dysplasia (Sakamoto et al., 1999).
Overexpression of FOSL1 increases ILD responding to TLR signaling (Takada et al., 2011). FOSL1 expression in the lungs mediates antifibrotic effects by modulating proinflammatory, profibrotic, and fibrotic gene expression, providing a potential biomarker for pulmonary fibrosis with poor prognosis and treatment options (Rajasekaran et al., 2012). E74-like ETS transcription factor 3 (ELF3) was upregulated by the inflammatory cytokines, interleukin-1β and TNF-α, in bronchial epithelial cell lines (Wu et al., 2008), playing essential functions in regulating epithelial cell differentiation during lung regeneration (Oliver et al., 2012). Nuclear receptor subfamily 4 group A member 1 (NR4A1), a transcription factor expressed in various tissues, was found to promote fibrogenesis by persistent stimulation with TGF-β1 and AKT-dependent phosphorylation (Zeng et al., 2018).
There are a few limitations in the present study. First, the sample size in the microarray dataset was limited. Second, findings of the in silico identification of DEGs and DE-miRNAs and their potential target genes lack experimental validation in vitro and in vivo. Finally, the associations between miRNA and mRNA expression profiles require further experimental validation. Future investigations with larger clinical sample sizes, along with optimized validation procedures, are needed.
Data Availability
Data supporting the results of the present investigation are available from the corresponding author upon reasonable request.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was financially supported by the National Natural Science Foundation of China (No. 81602004 and No. 81600259).