
Abstract
Multiple factors, including genetic and epigenetic fluctuations, have been linked to gastric cancer formation and progression. Cytosine methylation (5mC) has been recognized as a critical epigenetic mark in the mammalian genome. The recent discovery of 5-hydroxymethylcytosine (5hmC), which is generated by ten-eleven translocation (TET) enzymes, provides new perspectives to understand DNA methylation-related plasticity. In this study, we show that gastric tumors display significant loss of 5hmC. Using matched distant normal, peripheral, and tumor primary tissues, we performed genome-wide profiling of 5hmC and identified differentially hydroxymethylated regions (DhMRs) specifically associated with gastric tumors. Gene ontology analyses indicated that DhMRs (both loss of 5hmC and gain of 5hmC) were enriched among the genes involved in specific pathways. Interestingly, the binding motif of hypoxia-inducible factor 1 (HIF1) is enriched among both peripheral and tumor DhMRs, while the Myc-binding motif is specifically enriched among only tumor DhMRs. Tumor progression analyses revealed a unique set of DhMRs that correlate with tumor progression. These data together suggest that alteration of 5hmC could potentially contribute to the tumorigenesis of gastric tumors.
Introduction
Gastric cancer (GC) is one of the most common cancers and leading causes of global cancer mortality (Ferlay et al., 2015). Recently, a substantial number of studies have revealed that GC is a multistage pathological state (Tan and Yeoh, 2015). The formation and progression of GC can be triggered by various factors, including internal molecular/genetic changes and external environmental exposures (e.g., viral/bacterial infections, smoking, and diet) (Ajani et al., 2017).
Epigenetics is a term that defines heritable changes in gene expression, where the DNA sequence is not altered (Egger et al., 2004). In general, there are three well-studied epigenetic marks, including histone modification, DNA methylation, and noncoding RNA-mediated silencing (Padmanabhan et al., 2017). Disrupting any of the abovementioned epigenetic systems can result in dysregulated gene expression and, thereafter, induce a number of multisystem disorders and neoplasia. For example, previous studies have identified several key genetic alterations that are linked to gastric malignancy, including mutations in the chromatin modifier gene AT-rich interaction domain 1A (ARID1A) and amplifications in human epidermal growth factor receptor 2 (HER2), fibroblast growth factor receptor 2 (FGFR2), and mesenchymal–epithelial transition (MET) (Ajani et al., 2017). Moreover, the recent histone modification study in primary GC revealed a pervasive reprogramming of the gastric superenhancer landscape during tumorigenesis (Ooi et al., 2016).
In addition to histone modifications, cytosine methylation (5mC) can influence transcriptional states by modifying DNA-protein interactions (Jaenisch and Bird, 2003). Dysregulation at the epigenetic level, including hypermethylation on promoter CpG islands, is a commonly recognized molecular cause of human neoplasia. The hypermethylation on the mismatch repair gene human mutL homolog 1 (hMLH1) promoter region is the main underlying mechanism for microsatellite instability in GC (Keller et al., 1996). Another example is the methylation level on the p16 (cyclin-dependent kinase inhibitor 2A) gene. It has been reported that although mutation of the p16 gene is infrequent, p16 hypermethylation is common in GC with a higher incidence in the intestinal type, suggesting a vital role of DNA methylation-mediated p16 gene inactivation in GC (Shim et al., 2000).
More recently, a novel cytosine modification, 5-hydroxymethylcytosine (5hmC), was discovered. The ten-eleven translocation (TET) family of dioxygenases oxidizes 5mC to generate 5hmC (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009). Subsequent studies demonstrated that TET proteins can further oxidize 5hmC to 5-formylcytosine and 5-carboxylcytosine, which are recognized and excised by mammalian DNA glycosylase thymine-DNA glycosylase (TDG) and subsequently converted to cytosine through base excision repair (Cortazar et al., 2011; Cortellino et al., 2011; He et al., 2011; Ito et al., 2011; Maiti and Drohat, 2011; Pfaffeneder et al., 2011; Zhang et al., 2012), resulting in active DNA demethylation in mammals. TET-mediated epigenetic modification is critical in embryonic stem cell (ES) maintenance, normal myelopoiesis, myeloid leukemia, and gliomas, pointing to the biological importance of 5hmC modification in regulation of ES pluripotency, tissue homeostasis, and disease pathogenesis (Ito et al., 2010; Ko et al., 2010; Dawlaty et al., 2011; Gu et al., 2011; Koh et al., 2011; Moran-Crusio et al., 2011; Williams et al., 2011; Xu et al., 2011; Doege et al., 2012; Lian et al., 2012; Tan and Shi, 2012). In particular, 5hmC abundance is significantly reduced in many types of human cancers, such as melanoma (Lian et al., 2012), prostate, breast, and colon cancers (Haffner et al., 2011), hepatocellular carcinoma (Liu et al., 2013), and especially in central nervous system tumors (Jin et al., 2011). However, genome-wide analyses of 5hmC in GC tissues have not been conducted.
In this study, using matched distant normal, peripheral, and tumor primary tissues from six patients, we show that the loss of 5hmC is specifically associated with gastric tumors. Genome-wide profiling of 5hmC identified differentially hydroxymethylated regions (DhMRs) specifically associated with GC. Gene ontology (GO) analyses indicated that DhMRs (both loss of 5hmC and gain of 5hmC) were enriched among the genes involved in specific pathways. Interestingly, the binding motif of hypoxia-inducible factor 1 (HIF1) is enriched among both peripheral and tumor DhMRs, while the Myc-binding motif is specifically enriched among tumor DhMRs. Furthermore, we performed tumor progression analyses and identified a unique set of DhMRs that correlate with tumor progression. Finally, we observed significant overlap between DhMRs and previously identified superenhancers. These data together suggest that alteration of 5hmC could potentially contribute to the tumorigenesis of gastric tumors.
Materials and Methods
Sample collection
The protocol for this study was approved by the Ethics Committee of The Second Hospital of Jilin University (20160208). Patients recruited for this study provided informed consent to participate, and written informed consents were obtained from all the tissue donors of this study (Supplementary Table S1). Sets of fresh gastric tissues were collected from six patients, including gastric tumor (tumor/T), gastric tissue adjacent to the tumor (peripheral/P), and distant normal gastric tissue (normal/N). Samples were stored at −80°C until DNA was extracted.
Dot blot assay
Genomic DNA was extracted by using phenol–chloroform and then quantified by NanoDrop 2000 (Thermo). After being denatured in 1M NaOH for 2 h, 100–300 ng of genomic DNA was spotted onto a charged nylon-based membrane at room temperature for 30 min and then baked at 75°C for 1 h. The membrane was blocked with 5% nonfat milk for 1 h and washed in phosphate-buffered saline (PBS) three times. The blot was then incubated with rabbit polyclonal anti-5hmC antibody (Active Motif; 1:10,000) at 4°C overnight. On the following day, the blot was washed in PBS three times and incubated with peroxidase-conjugated anti-rabbit IgG secondary antibody for 1 h. The signal was visualized using the ECL Plus system (Amersham Pharmacia Biotech) and quantified.
5hmC-specific chemical labeling, affinity purification, and sequencing
Genomic DNA was used for 5hmC genome-wide profiling. 5hmC enrichment was performed using a previously described procedure with an improved selective chemical labeling method (Song et al., 2011). DNA libraries were generated following the Illumina protocol for Preparing Samples for ChIP Sequencing of DNA (Part# 111257047 Rev. A) using 25–50 ng of input genomic DNA or 5hmC-captured DNA to initiate the protocol.
Bioinformatic data analyses
Processing of sequencing data was performed as previously described (Wang et al., 2013). Briefly, FASTQ sequence files from biological replicates were concatenated and aligned to the Homo sapiens reference genome (GRCh37/hg19) using Bowtie 0.12.6, keeping only unique, nonduplicate genomic matches with no more than two mismatches within the first 25 bp. Hydroxymethylation levels on each CpG site were evaluated by read counts. To determine differentially hydroxymethylated CpG sites/regions, we used edgeR (version: edgeR_3.8.6) to normalize read counts, estimate dispersion, and perform differential tests; p-values smaller than 0.01 are considered significant to call DhMRs.
Motif detection within DhMRs was performed with Homer. DhMRs were resized to 50-bp windows fixed at the center. Homer (v 4.8.3) was used to scan DhMRs for motifs using findMotifsGenome.pl with a standard background (random genomic sequences sampled according to the GC content of peak sequences) and parameters (mask repeats/lower case sequence, using a 200-bp fragment for motif finding).
GO analyses were performed as previously described using the Database for Annotation, Visualization and Integrated Discovery (DAVID) Bioinformatics Resources 6.7 Functional Annotation Tool. Gene sets were identified by joining subsets of DhMRs with RefSeq tables obtained from the UCSC genome browser tables.
Statistical analyses
GraphPad Prism software was used for statistical analyses. Either a t-test or one-way analysis of variance (ANOVA) was performed to evaluate differences between groups. All results are expressed as mean ± standard error of the mean; p-values <0.05 were considered statistically significantly different.
Data availability
The sequencing data are archived at Gene Expression Omnibus under accession number GSE134927.
Results
5hmC is significantly reduced in gastric tumors
Previous studies have found 5hmC abundance to be significantly reduced in several different types of human cancers. To explore the role of 5hmC in GC, we collected six sets of fresh gastric tissues, including gastric tumor (tumor/T), gastric tissue adjacent to the tumor (peripheral/P), and distant normal gastric tissue (normal/N) (Supplementary Table S1). We first examined 5hmC abundance by dot blotting using an anti-5hmC antibody. Pairwise comparisons revealed that gastric tumor tissue (T) showed a significant reduction in 5hmC compared with normal gastric tissue (N) (*p < 0.001; Fig. 1A), with an average of 0.35-fold 5hmC relative to normal tissue. No significant difference was observed between peripheral tissue (P) and normal tissue (N), suggesting that reduction of 5hmC is a hallmark of GC.
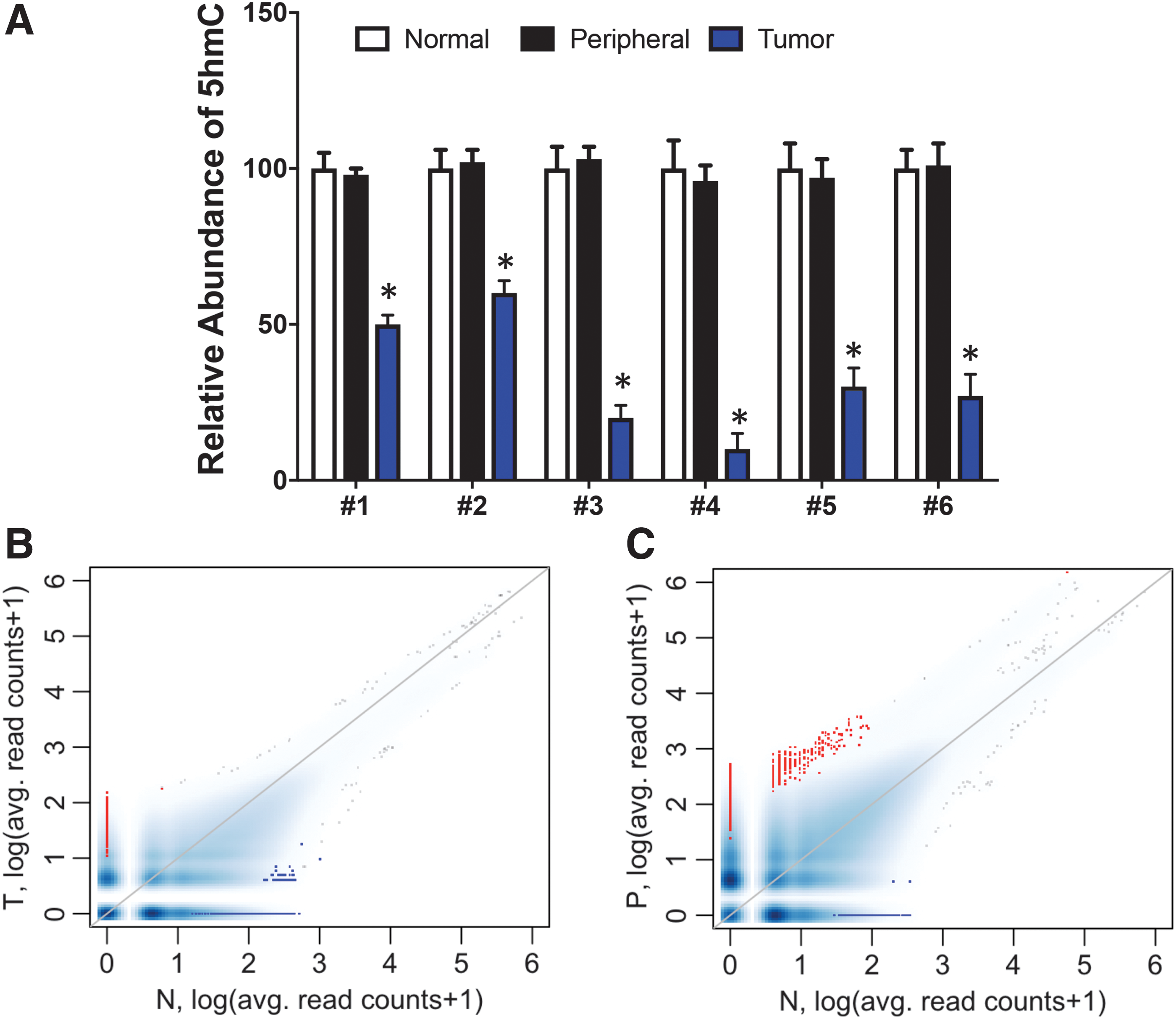
5hmC is significantly reduced in gastric tumors.
Genome-wide 5hmC profiling in gastric tumors
To understand the alteration of 5hmC in gastric tumors, we investigated 5hmC genome-wide distribution in all three tissue types using a chemical labeling and affinity purification method coupled with high-throughput sequencing (Song et al., 2011). First, global 5hmC levels were determined by counting and plotting normalized 5hmC mapped reads in each 10-kb binned human genome among the three sets of tissues. Bins with lower 5hmC reads in tumor or peripheral tissue compared with normal tissue are shown in blue, whereas bins with higher 5hmC reads in tumor or peripheral tissue compared with normal tissue are shown in red. In the comparison of tumor tissue with normal tissue, we found an overall higher number of bins in blue, indicating a general reduction of global 5hmC in tumor tissue (Fig. 1B, blue vs. red), which is consistent with our dot blot analyses. Intriguingly, there were more red bins than blue bins in the comparison of peripheral tissues with normal tissue (Fig. 1C), suggesting that the 5hmC abundance could potentially increase in peripheral tissues; however, we did not detect a significant difference by dot blot.
Identification and characterization of DhMRs
To identify the differentially hydroxymethylated CpG sites, edgeR (version: edgeR_3.8.6) was used to normalize read counts, estimate dispersion, and perform differential tests. Tumor, peripheral, and normal tissues from the same individual were grouped together to adjust for any within-subject effects (Supplementary Table S2). Sites with p-values smaller than 0.01 were considered significantly different DhMRs and included in the downstream analyses. The genome-wide distribution of DhMRs is shown in Figure 2A. Overall, 17,318 5hmC-containing genomic regions display reduced 5hmC, while 1344 loci show increased 5hmC in tumor versus normal tissues, consistent with the overall reduction of 5hmC abundance observed earlier. Interestingly, in peripheral tissues versus normal tissues, more genomic loci display increased 5hmC signals (4795) than reduced signals (1876) (Table 1, Fig. 2B).
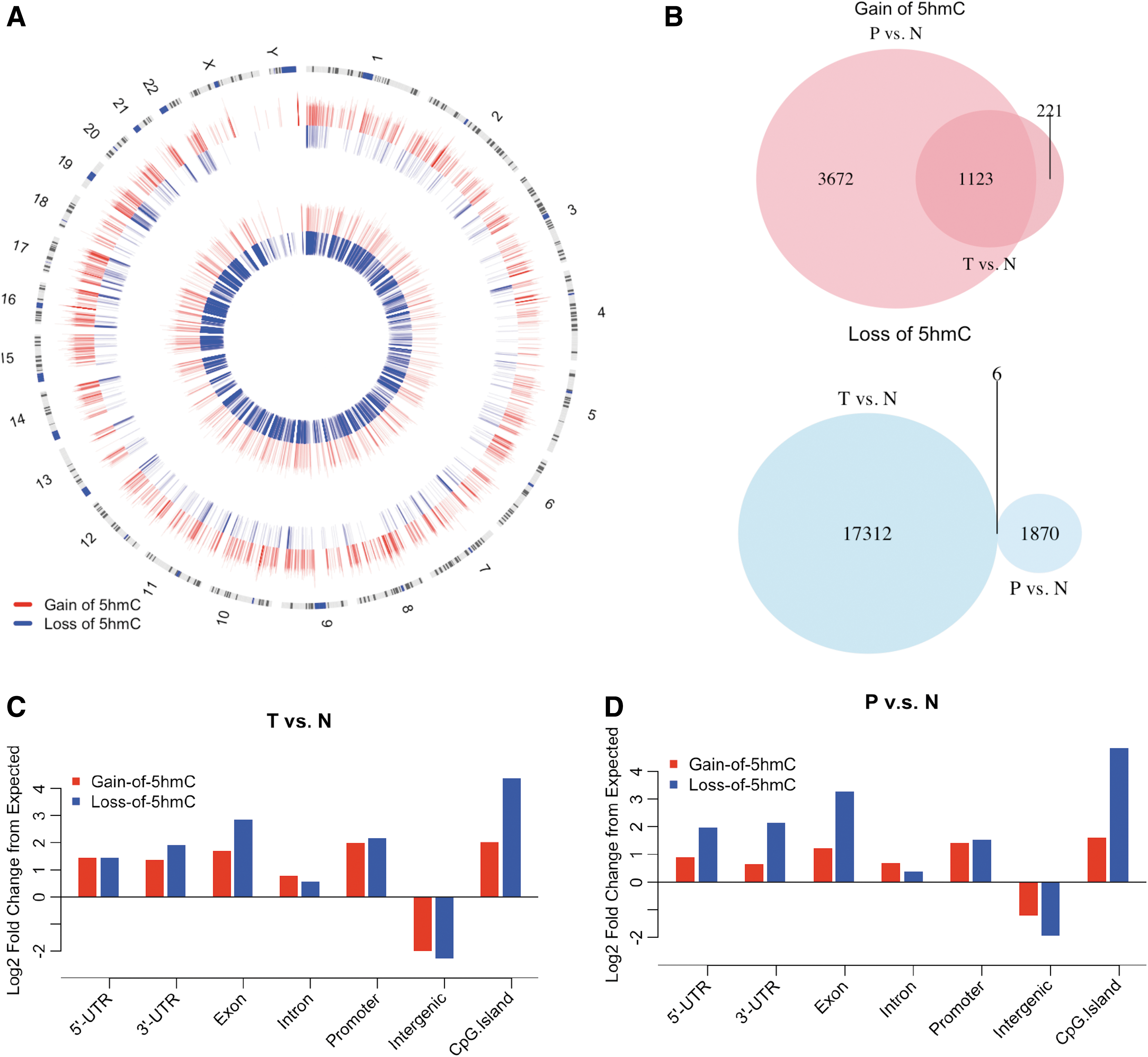
Identification and characterization of DhMRs.
Number of Differentially Hydroxymethylated Regions
5hmC, 5-hydroxymethylcytosine; P/N, ratios between P and N; T/N, ratios between T and N; T-P-N progression, tumor–peripheral–normal tissue progression.
To investigate the relationship of DhMRs between these two comparisons, we overlapped the regions with gain of 5hmC or loss of 5hmC from the two comparison groups. Interestingly, 1123 of the 1344 (83.5%) DhMRs from the ratios between T and N (T/N) gain of 5hmC DhMRs overlapped with the ratios between P and N (P/N) gain of 5hmC DhMRs, suggesting similar 5hmC changes in both tumor and peripheral tissues. However, the loss of 5hmC regions from these two groups were very distinct: only six common loss of 5hmC regions were identified as overlapping (0.3% of P/N and 0.03% of T/N) (Fig. 2B).
We next characterized the genome-wide distributions of DhMRs and evaluated their enrichment or depletion on featured genomic regions by comparing with expected values. DhMRs were enriched on intragenic regions (e.g., exons, UTRs, and promoters) (Fig. 2C, D, log2-fold change from expected >0), while they were depleted in intergenic regions. The results supported the previous observations suggesting that intragenic 5hmC may be associated with gene regulation (Song et al., 2011).
Aberrant 5hmC changes in gastric tumors and peripheral tissue influence the core biological pathways and potential binding affinity of selective transcription factors
To understand whether dynamic 5hmC changes have impact on the relevant biological processes, we mapped these GC-associated DhMRs to the hg19 human reference sequence and further subjected these genes to GO pathway analysis (Huang da et al., 2009). Interestingly, several biological processes, such as the phosphate metabolic process, protein amino acid phosphorylation, and intracellular signaling cascade, were affected by gain of 5hmC in both peripheral and tumor tissues (Fig. 3A, C). GO analysis also revealed unique pathways associated with either peripheral or tumor tissues. For instance, the pathway related to Ras protein was uniquely featured in peripheral tissue, but not in tumor tissue. Among the loss of 5hmC loci, the overlap between peripheral and tumor tissues was relatively small compared with those with gain of 5hmC (e.g., Ras protein transduction) (Fig. 3B, D). The loss of 5hmC loci in peripheral tissues were enriched with genes involved in programmed cell death, while the loss of 5hmC loci associated with tumor tissues were enriched with genes involved in cytoskeleton organization and intracellular signaling. These observations suggest unique 5hmC alterations associated with peripheral and tumor tissues, reflecting different stages of tumorigenesis.
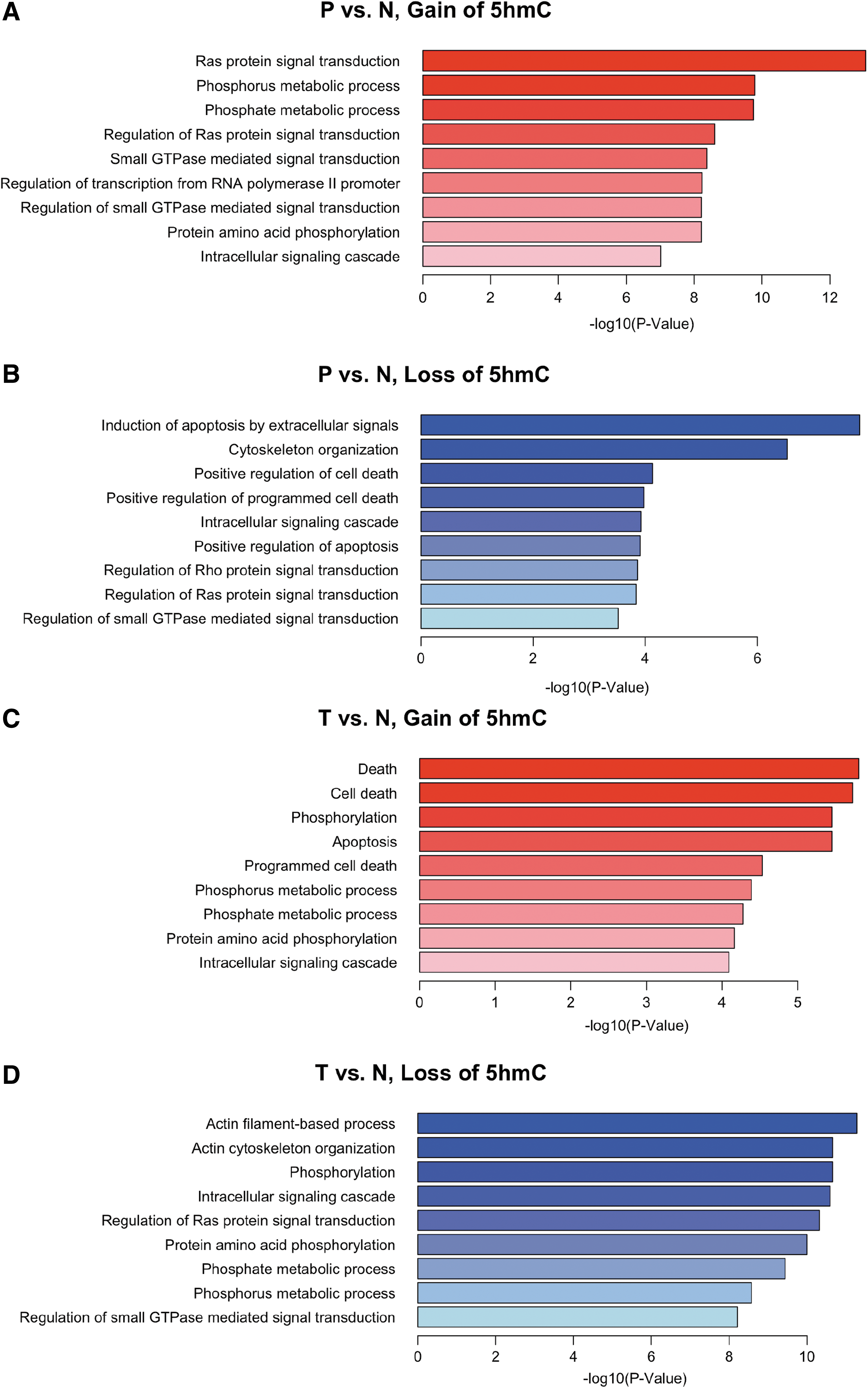
GO analysis with gain and loss of 5hmC. Genes with DhMRs on their gene body were defined as DhMR-associated genes. The DAVID analysis was used for the DhMR-associated genes in P/N
Given that many transcription factors are sensitive to DNA covalent modification dynamics, we employed motif search algorithms to comprehensively predict the potential transcription factor binding sites on the DhMRs that we identified. Interestingly, the binding motif of HIF1, a critical transcription factor responsible for controlling the transcriptome in response to normoxia and hypoxia, was enriched in both peripheral and tumor DhMRs (Fig. 4A, B). In addition, specifically among tumor DhMRs, Myc (both c-Myc and n-Myc) binding sites were enriched. These results suggest that the altered 5hmC modifications could potentially alter the binding of both HIF1 and Myc during gastric tumorigenesis.

Motif search analysis of DhMRs. Motif search using DhMRs identified in P/N
Tumor progression analyses
Since we have three distinct tissues, tumor, peripheral, and distant normal tissues, we performed a tumor–peripheral–normal tissue progression test. This analysis aimed to identify regions that displayed a gradual gain or loss of 5hmC along the progression of tissue changes. In this analysis, T, P, and N tissues were coded as the three stages of a continuous progression and quantified as levels 2, 1, and 0, respectively. At the differential calling step, the stages were used as the alternative column to group in the design matrix while adjusting for individual effects. Our progression analysis revealed a distinct set of DhMRs (74 loci with gain of 5hmC and 3481 loci with loss of 5hmC; Table 1). The changes of 5hmC within these regions are shown in Figure 5A as a heatmap, which correlates with disease progression.
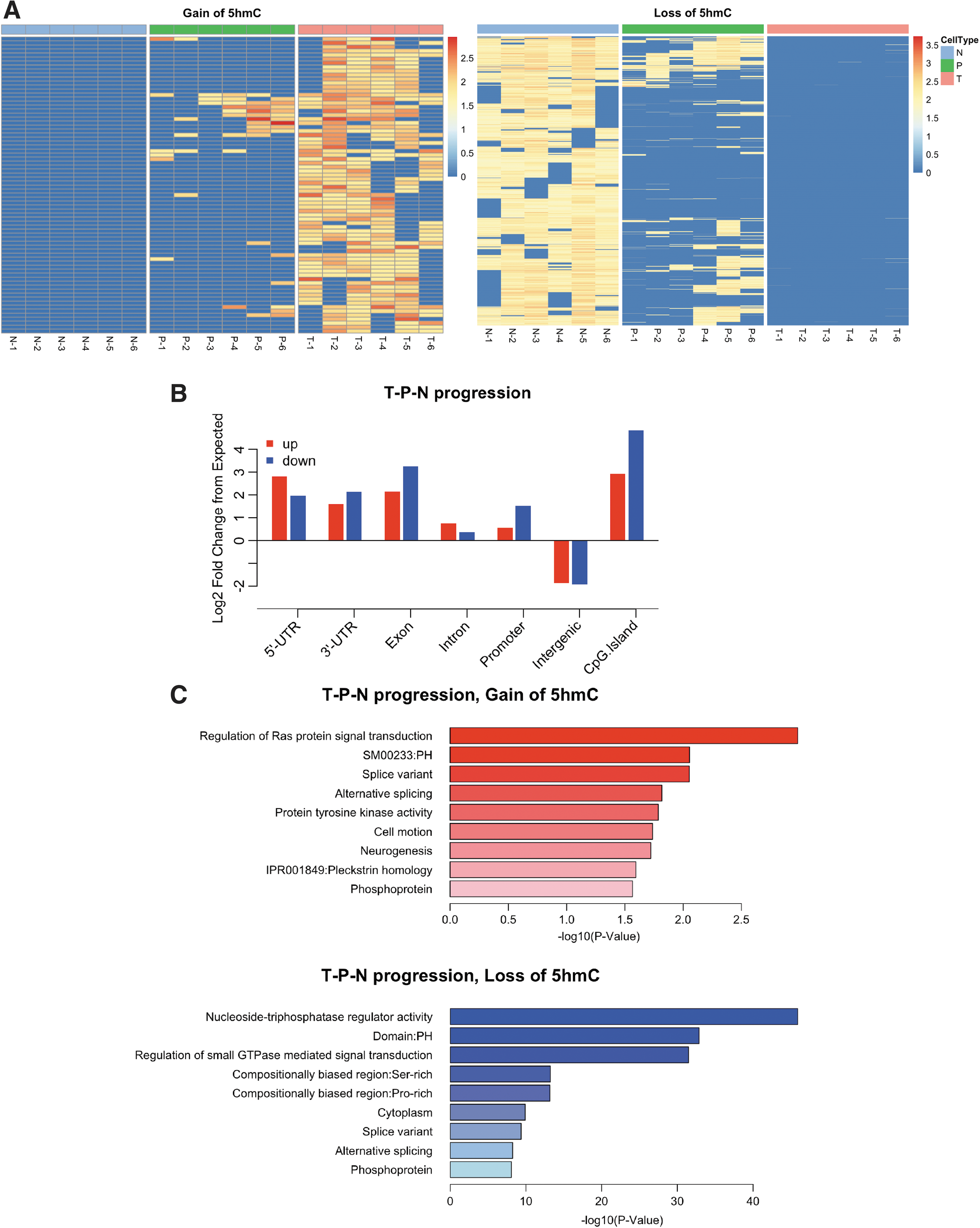
Tumor progression analyses.
Genomic features associated with this unique set of DhMRs were similar to the DhMRs of both peripheral and tumor tissues (Fig. 5B compared with Fig. 2C, D). However, the functions of DhMRs, as characterized in functional enrichment of GO terms, are distinct from the previous two groups of DhMRs (Fig. 5C compared with Fig. 3). In particular, among the loci with loss of 5hmC in the progression analyses, genes involved in splicing and nucleoside regulators were uniquely enriched.
DhMRs overlap with the previously identified superenhancers
Superenhancers refer to enhancer clusters occurring in close proximity, are larger in size than typical enhancers, exhibit higher transcription factor binding densities, and are strongly associated with key cell identity regulators. Superenhancers are enriched in disease-associated genetic variants and are acquired by cancer cells at key oncogenes. By analyzing more than one hundred epigenomic profiles from primary GC, normal gastric tissues, and cell lines, earlier works have identified superenhancers associated with GC tumorigenesis and revealed a genome-wide reprogramming of the superenhancer landscape during GC tumorigenesis (Ooi et al., 2016). To correlate the 5hmC dynamics with superenhancers, we examined the change of 5hmC at the top 100 superenhancers identified in the previous study and performed enrichment analysis. The analyses were performed by comparing the following two ratios: (1) DhMRs overlapping with superenhancers/all DhMRs and (2) superenhancers/the whole genome. The fold change of ratio (1) versus (2) is shown in Figure 6 (in log2 scale). In addition, binomial tests were performed to evaluate the significance of enrichment using a one-sided null hypothesis and the probability of one nucleotide being inside the superenhancer, that is, ratio (2), as the percentage of success. We observed significant enrichment of the identified superenhancers among the DhMRs of both peripheral and tumor tissues.
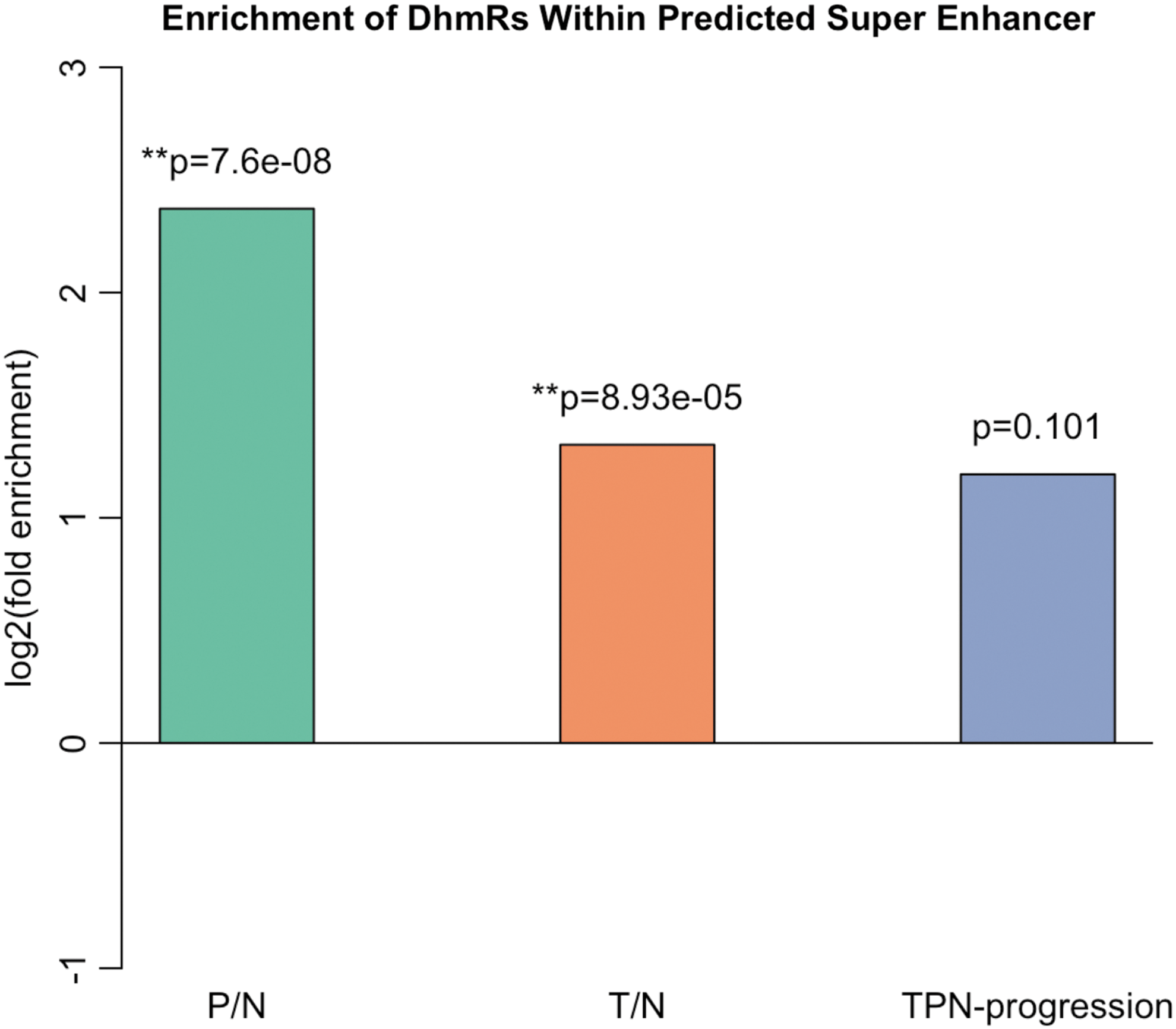
Overlaps between DhMRs and superenhancers. Enrichment analysis was performed by examining the 5hmC change at the top 100 superenhancers identified in the previous study. Analyses were performed by comparing the following two ratios: (1) the ratio between DhMRs overlapping with superenhancers and the whole range of DhMRs and (2) the ratio between superenhancers and the whole genome. The fold change of ratio (1) versus (2) is highest in P/N, followed by T/N, and least in T-P-N progression. A binomial test was performed to evaluate the significance of enrichment using a one-sided null hypothesis and the probability of one nucleotide being inside the superenhancer, that is, ratio (2), as the percentage of success. **the enrichment is significant.
Discussion
In the past decade, increasing evidence has suggested that epigenetic regulation plays a vital role in the initiation and progression of human cancer, including gastric cancer (Jones and Baylin, 2002). Not merely a transient intermediate in the DNA active demethylation process, 5hmC is reported to be enriched and stable in mammalian tissues and recognized by specific 5hmC-binding proteins (Szulwach et al., 2011; Spruijt et al., 2013). In addition, it is known that 5hmC has biological functions in a number of developmental and neurological diseases and cancers (Szulwach et al., 2011; Lian et al., 2012; Cheng et al., 2015). Thus, 5hmC could potentially contribute to the tumorigenesis of gastric tumors. In the present study, for the first time, we profiled the genome-wide 5hmC in matched distant normal, peripheral, and tumor primary tissues from GC patients, and our results show a specific association between the loss of 5hmC and gastric tumor tissues. GO analyses indicated that DhMRs (both loss of 5hmC and gain of 5hmC) were enriched among the genes involved in a number of selective pathways. Interestingly, the binding motif of HIF1 is enriched among both peripheral and tumor DhMRs, while the Myc-binding motif is specifically enriched among tumor DhMRs. The tumor progression analyses identified a unique set of DhMRs that correlate with gastric tumor progression. In addition, we observed significant overlap between DhMRs and previously identified superenhancers. These data together suggest that the alteration of 5hmC could potentially contribute to tumorigenesis and progression of gastric tumors.
As the first epigenetic mark that was linked to tumorigenesis (Feinberg and Vogelstein, 1983), DNA methylation provides a stable gene-silencing mechanism, and it is commonly known that hypermethylation in promoter regions causes inactivation of certain tumor suppressor genes (Kulis and Esteller, 2010). In addition, studies have found that hypomethylation could also result in activation of genes that are important in cancer (Feinberg and Tycko, 2004). It is interesting that our tumor progression analyses suggest that the loss of 5hmC is more significantly correlated with gastric tumor progression. The GO analysis revealed that the loss of 5hmC loci in peripheral and tumor tissues is distributed in genes involved in distinct pathways, with the exception of a few well-known pathways that are associated with tumorigenesis of gastric tumors (e.g., Ras protein signal transduction). It has been reported that an oncogene, BRAF (serine/threonine protein kinase B-Raf), encoding an RAF protein in the downstream of the RAS pathway, is somatically mutated in many human cancers, including GC (Davies et al., 2002). This suggests that aberration of the RAS pathway could contribute to the pathogenesis of stomach cancer. Therefore, the loss of 5hmC may result in dysregulation of the RAS genes and contribute to gastric tumor progression.
It was once considered that only protein-coding DNA (about 2% of the hg19) contained valuable information, while the rest of the genomic noncoding DNA was considered junk DNA. However, more recent genome-wide studies have revealed an understanding of the hidden information in the human genome (Consortium, 2012), where more than the expected number of noncoding regions have been found to encode regulatory information. For example, it has been reported that levels of a long noncoding RNA, H19, were substantially upregulated in cells and tissues of GC when compared with normal controls, resulting in partial p53 inactivation (Yang et al., 2012). Previous global profiling of chromatin marks that are associated with distinct regulatory elements has revealed the alterations of hundreds of epigenomic promoters and predicted enhancers in GC (Muratani et al., 2014; Ooi et al., 2016; Qamra et al., 2017). In this study, we overlapped the top 100 superenhancers identified in previous studies and performed enrichment analyses. Many of the DhMRs of both peripheral and tumor tissues overlap with predicted superenhancers, suggesting that the dynamic change of 5hmC may influence the expression of noncoding regions and adjacent genes. It will be important to investigate the roles of 5hmC in our identified intergenic DhMRs in GC patients.
In summary, we profiled the genome-wide distribution of 5hmC in matched distant normal, peripheral, and tumor primary tissues from GC patients. Our results showed a specific association between the loss of 5hmC and gastric tumor tissues. The DhMRs located within genes associated with a number of selective pathways and were enriched with motifs that are involved in gastric tumorigenesis. In addition, we observed significant overlap between DhMRs and previously identified superenhancers. These data together suggest that the alteration of 5hmC could potentially contribute to the tumorigenesis of gastric tumors.
Authors' Contributions
X.W. and P.J. designed the experiments. H.L., T.X., Y.C., M.H.J., M.Y.C., and E.G.A. conducted all experiments. X.W. and P.J. wrote the article. T.X., Y.C., M.H.J., M.Y.C., and E.G.A. performed the data analysis and discussed the results. All authors read and approved the final article.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported, in part, Bethune Plan Research Project of Jilin University [470110000694 to X.W.] by the National Institutes of Health [NS051630, NS091859, and NS097206 to P.J.].
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.