
Abstract
Polo-like kinase 1 (PLK1) is a ubiquitous serine/threonine protein kinase. It is reported to be involved in the occurrence and progression of various human cancers. In the present study, we explored the role and molecular mechanism of PLK1 in the proliferation of osteosarcoma (
Introduction
Osteosarcoma (OS) is a commonly seen cancer with bone malignancies that occurs in teenagers and adolescents (Misaghi et al., 2018), accounting for about 5% of pediatric tumors (Durfee et al., 2016; Omer et al., 2017). OS is derived from mesenchymal cells, and its rapid tumor growth is due to the direct or indirect formation of tumor-like tissue and bone tissue (Hu et al., 2016; Liu et al., 2017). However, the molecular biological mechanism of OS still has not been clearly described.
Reversible protein phosphorylation regulated by protein kinases is the most basic molecular regulation mechanism in eukaryotes. Polo-like kinase 1 (PLK1) is a ubiquitous serine/threonine protein kinase with highly conserved structure and function (Lu et al., 2013). It plays an important role in cell cycle progression (Yamaguchi et al., 2009), centromere maturation, cytoplasmic separation in mitosis, and maintenance of genomic stability (Elez et al., 2003; Bucur et al., 2014). It has been reported that PLK1 plays an important role in the occurrence and development of various tumors, and it is also recognized as an important target for tumor treatment (Driscoll et al., 2014; Raab et al., 2018). In
PLK1 participates in the cell growth and invasion of renal cancer cells (Zhang et al., 2013), colorectal cancer cells (Han et al., 2012), and human esophageal cancer cells (Bu et al., 2008). PLK1 overexpression is detected in various human cancer cells, and the pathogenic mechanism has also been initially discussed in various cancers. For instance, downregulation of PLK1 increased drug sensitivity of breast cancer cells in vitro and in vivo (Spankuch et al., 2006). In nonsmall cell lung cancer, PLK1 inhibitor BI-6727, in combination with radiation, significantly decreased tumor growth. Interference with PLK1 enhanced radiosensitization by modulating DNA repair proteins, including DNA-dependent protein kinase (DNAPK) and topoisomerase II alpha (TOPO2A) (Yao et al., 2018). Phosphorylation of PLK1-Ser99 is dependent on the PI3K/Akt pathway, which is required for metaphase–anaphase transition. Moreover, cell apoptosis and proliferation of pancreatic cancer cells were regulated by the PI3K/Akt pathway through PLK1 (Mao et al., 2016).
Several PLK1 inhibitors are widely used in the study and clinical treatment of human cancers. For example, the PLK inhibitor TAK-960 is reported to possess antitumor activity against colorectal cancer (Klauck et al., 2018). BI2563, a potent and selective inhibitor of PLK1, is a widely used dihydropteridinone compound in preclinical and clinical studies. It inhibits PLK1 by binding to the ATP-binding site and cross-suppresses PLK2 and PLK3 simultaneously. It has been found that BI2536 and cisplatin synergistically suppress cell proliferation of gastric cancer cells (Lian et al., 2018). In the present study, we chose BI2563 as the positive control PLK1 inhibitor to treat
Materials and Methods
Cell lines
The human
Reagents
The 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) reagent was obtained from Sigma, Inc. (Saint Louis, MO). Doxorubicin (Cat. No.: D1515) was obtained from Sigma-Aldrich Corporation. The PLK1 inhibitor BI2536 was purchased from Selleck (Cat. No.: S1109). The following were obtained from Sigma: human kinase PLK1 siRNA1 (Cat. No.: SIHK1741), PLK1 siRNA2 (Cat. No.: SIHK1742), MISSION® siRNA universal negative control #1 (Cat. No.: SIC001), MISSION microRNA mimic hsa-miR-4779 (Cat. No.: HMI1866), MISSION miRNA negative control 1 (Cat. No.: HMC0002), MISSION synthetic microRNA inhibitor hsa-miR-4779 (Cat. No.: HSTUD1866), MISSION synthetic microRNA inhibitor-negative control (NCSTUD001), and MISSION lenti-microRNA inhibitor hsa-miR-4779 (Cat. No.: HLTUD1866). PLK1 cDNA ORF Clone (Cat. No.: HG10676-ACG) and the pCMV3-N-GFPSpark Control Vector (Cat. No.: CV027) were purchased from Sino Biological Corporation.
MTT assay
Cell viability was determined by MTT assay. In one experiment, MG63/Dox cells were transfected with PLK1 siRNA1, PLK1 siRNA2, or negative control siRNA for 24, 48, and 72 h. Cell viability was determined by MTT assay. In another experiment, MG63/Dox cells were treated with increasing concentrations of BI2536 for 24, 48, or 72 h. The concentrations of BI2536 were 5, 25, and 100 nM. Four hours before treatment, 10 μL of 5 mg/mL MTT reagent was added to the culture medium, and the absorbance of each well in a 96-well plate was read at 490 nm.
Cell transfection
MG63 cells and MG63/Dox cells at a concentration of 1 × 105 cells per well were plated into a 24-well plate and cultured overnight for adherence to the plate. Cells were then transfected with PLK1 siRNA1, PLK1 siRNA2, or negative control siRNA for an indicated time, such as 24, 48, and 72 h. The PLK1-specific siRNAs were transfected using Lipofectamine 2000 (ThermoFisher, MA) according to the manufacturer's protocol.
Flow cytometry assay
MG63/Dox cells were plated into six-well plates and transfected with PLK1 siRNA1 and negative control siRNA for 48 h. The cell cycle was analyzed by flow cytometry using the propidium iodide staining method. In brief, the single-cell suspension of 1 mL at a density of 2 × 106 cells/mL was prepared. Cells were fixed with 75% ethanol at 4°C for 1 h, centrifuged at 1000 rpm for 5 min, and resuspended with 1 mL phosphate-buffered saline (PBS). Then, 100 μL of 200 μg/mL RNase A was added and cultures were incubated for 30 min at 37°C. Finally, 100 μL of 1 mg/mL propidium iodide was added and incubated for 5 min at room temperature.
β-Galactosidase assay
Cell senescence was detected by β-galactosidase (senescence-associated β-galactosidase [SA-β-gal]) activity as described (Chou et al., 2016). In brief, MG63 cells were transfected with PLK1 siRNA1, PLK1 siRNA2, or negative control siRNA for 72 h. MG63 cells were fixed and stained with X-gal and washed three times with PBS buffer. Cells were photographed using an Olympus IX51 inverted microscope.
Antibodies
The following primary antibodies were used for immunoblotting: PLK1 (Cat. No.: 101119-T02, 1:1000; Sino Biological); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cat. No.: ab8245, 1:1000; Abcam); β-actin (Cat. No.: sc-8432, 1:1000; Santa Cruz Biotechnology); caspase-3 (Cat. No.: ab13585, 1:2000; Abcam); and poly ADP-ribose polymerase (PARP) (Cat. No.: 9542, 1:5000; Cell Signaling Technology). Secondary antibodies used were as follows: goat anti-rabbit IgG H&L HR (Cat. No.: ab205718, 1:10,000; Abcam) and goat anti-mouse IgG-HRP (Cat. No.: sc-2005, 1:10,000; Santa Cruz Biotechnology).
Western blot analysis
Cell lysates were prepared using RIPA buffer (Cat. No.: P0013K; Beyotime, Shanghai, China), and 30 μg/lane of total proteins were subsequently separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). All proteins were transferred into polyvinylidene difluoride (PVDF) membrane, and the membrane was blocked with 5% solution of skimmed milk powder for 1 h at room temperature. The membrane was incubated with the indicated primary antibodies at 1:1000 and incubated at 4°C overnight. The membrane was then washed three times with 1 × Tris-buffered saline +0.1% Tween 20 buffer, each time for 10 min. The corresponding secondary antibody (at 1:10,000 dilution) was incubated for 40 min at room temperature. The membrane was washed with 1 × Tris-buffered saline +0.1% Tween 20 buffer at room temperature three times again. The bands were visualized using enhanced chemiluminescence reagents (Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
The data in the present study were analyzed using the SPSS statistical package. All results are shown as the mean ± standard error of the mean. Results were analyzed using an independent samples t-test. p-values <0.05 was considered to indicate a statistically significant difference.
Results
PLK1 levels increase in Dox-resistant OS cell lines
Drug resistance is usually observed during cancer treatment. In the present study, we used a pair of human
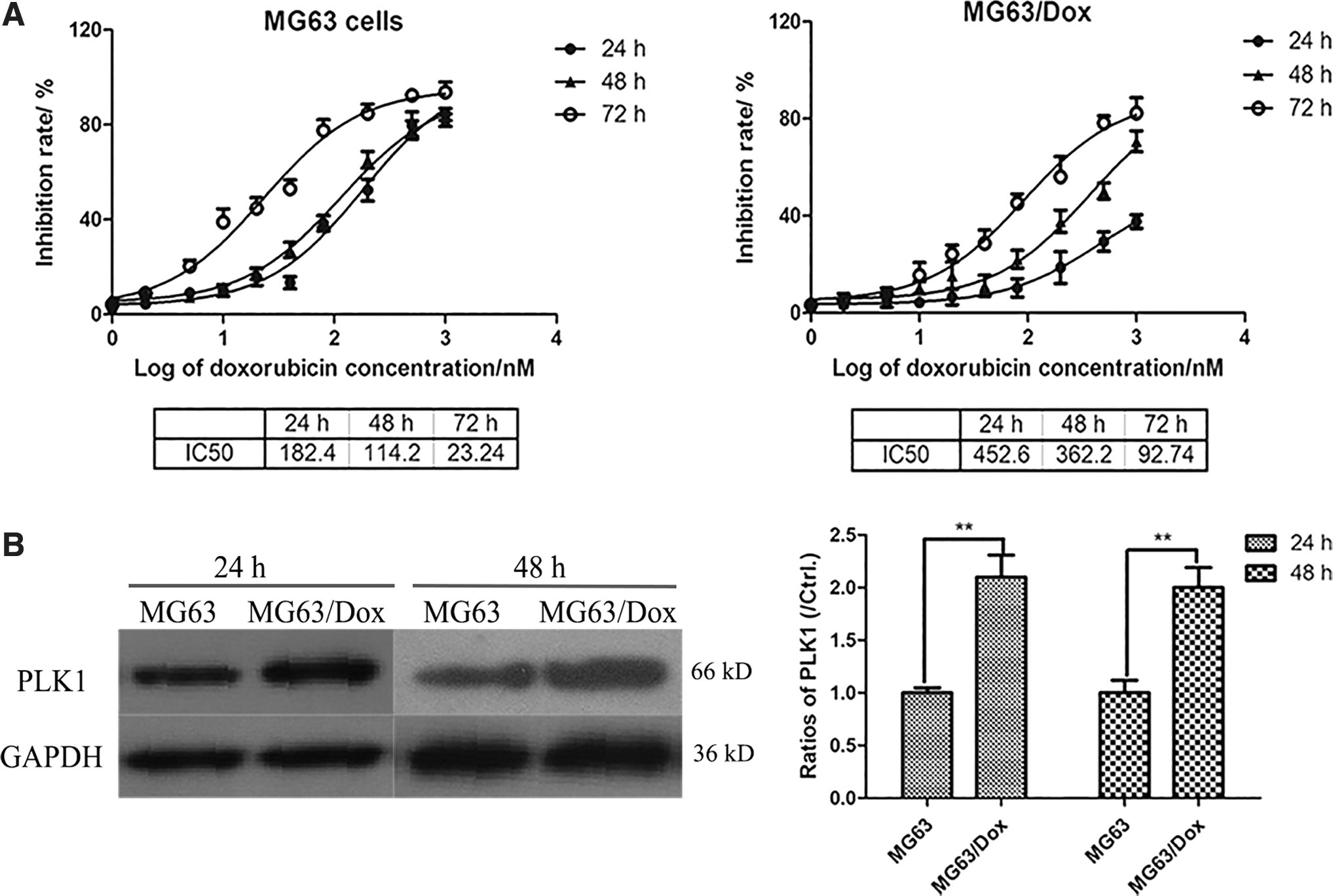
PLK1 levels are increased in Dox-resistant
To identify the role of PLK1 in chemotherapy of
Interference with PLK1 expression suppresses cell viability of Dox-resistant OS cells
To identify the role of PLK1 in proliferation of MG63/Dox cells, PLK1 siRNA1, PLK1 siRNA2, or negative control siRNA were used to transfect MG63 cells and MG63/Dox cells for 48 h. As depicted in Figure 2A, PLK1 was successfully suppressed by PLK1-specific siRNA1 or PLK1-specific siRNA2. Cell viability was determined by MTT assay, and the results demonstrated that cell proliferation was significantly suppressed more by PLK1 siRNA1- or PLK1 siRNA2-transfected MG63/Dox cells for 48 or 72 h than by negative control siRNA-transfected cells.

Inhibiting the expression of PLK1 suppresses cell viability of Dox-resistant
BI2536, a potent PLK1 inhibitor, was also used to treat MG63/Dox at 5, 25, and 100 nM for 48 h, and the level of PLK1 was determined by western blotting. As shown in Figure 2B, the level of PLK1 clearly decreased as the concentration of BI2536 increased. In addition, MG63/Dox cells were treated with 5 nM, 25 nM, and 100 nM for 24 h, 48 h, and 72 h, and the cell viability was determined by MTT assay. Cell viability was significantly inhibited by BI2536 in a dose-dependent manner.
Knockdown of PLK1 induces G2/M arrest and cell apoptosis in MG63/Dox cells
To examine whether transfection with PLK1 siRNA affected cell cycle progression of MG63/Dox cells, cell cycle analysis was performed by fluorescence-activated cell sorting (FACS) assay with the propidium iodide staining method. As shown in Figure 3A, the cell ratio reached 34.8% at G2/M phase in PLK1 siRNA-transfected MG63/Dox cells, which is higher than the ratio of 14.2% at G2/M phase in negative control siRNA-transfected MG63/Dox cells. All results showed that knockdown of PLK1 induced G2/M arrest in MG63/Dox cells.
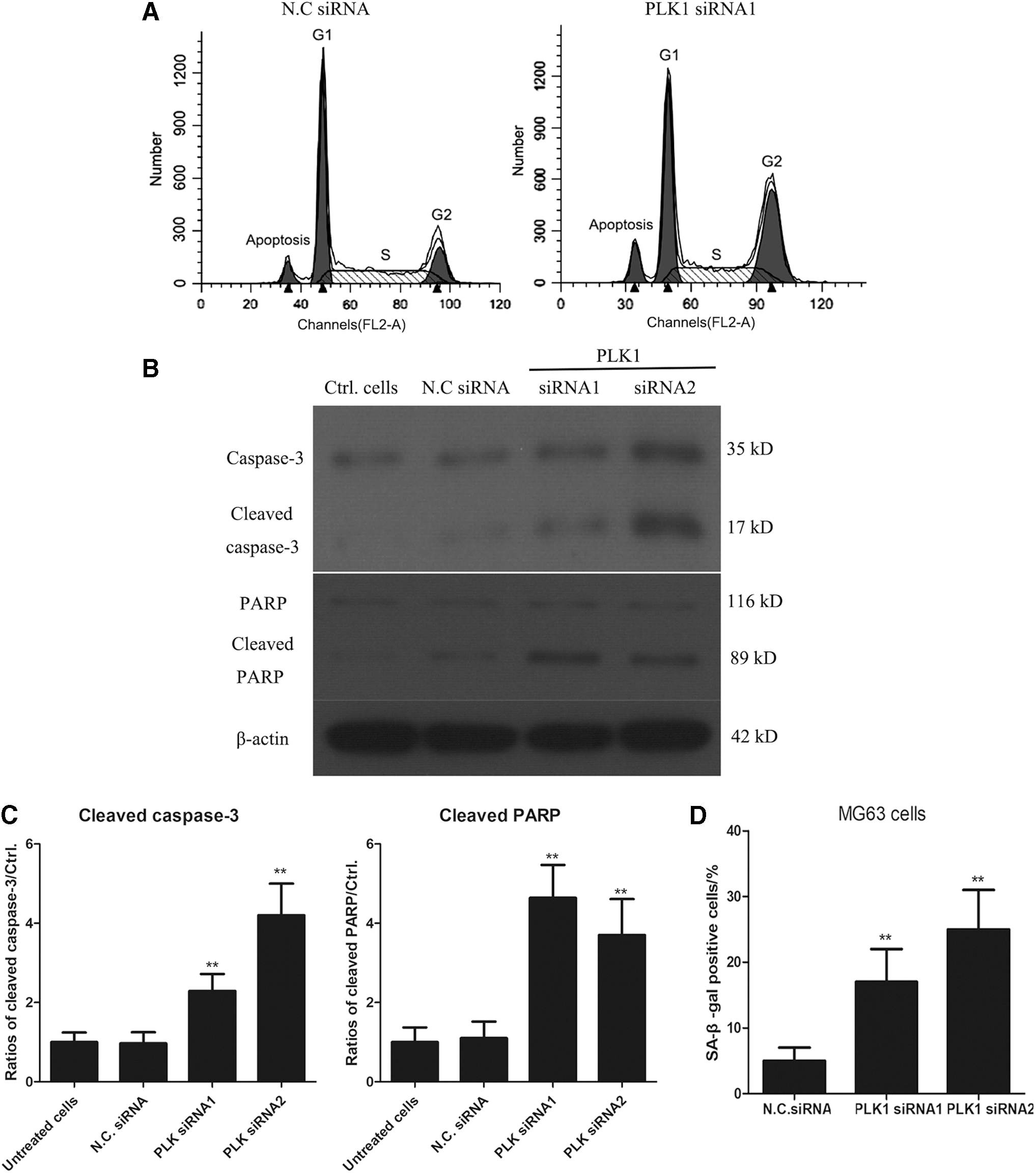
Knockdown of PLK1 induces G2/M arrest and cell apoptosis in MG63/Dox cells.
We then tested whether knockdown of PLK1 induced cell apoptosis of MG63/Dox cells. Interestingly, we found that the apoptosis rate in PLK1 siRNA-transfected MG63/Dox cells reached 15.3%, significantly higher than in negative control siRNA-transfected MG63/Dox cells (4.2%) as measured by cell cycle analysis (Fig. 3B). Levels of caspase-3 and cleaved caspase-3 were detected by western blotting. As shown in Figure 3B, in PLK1 siRNAs-transfected MG63/Dox cells, the levels of cleaved caspase-3 were obviously increased relative to negative control siRNA-transfected MG63/Dox cells. PARP cleavage is considered an important indicator of apoptosis and an indicator of caspase 3 activation. We also found that levels of PARP in PLK1 siRNA-transfected MG63/Dox cells were clearly increased compared to negative control siRNA-transfected MG63/Dox cells. The data suggest that knockdown of PLK1 induced cell apoptosis of MG63/Dox cells.
Interference with PLK1 promoted cell senescence of OS cells
In addition, the relationship of PLK1 and senescence of tumor cells was studied. The results showed a significant increase in SA-β-gal activity in PLK1 siRNA-transfected MG63 cells, suggesting that PLK1-KO induced senescence of OS cell lines, which was tested by SA-β-gal assay (Fig. 3D).
Has-miR-4779 regulates the expression of PLK1 in OS cell lines
MiRNAs are a class of short, noncoding RNAs that generally bind to the 3′-UTR of their target mRNAs to regulate gene expression posttranscriptionally. In the present study, TargetScan analysis was performed to predict the miRNAs that regulate the translation of PLK1. As shown in Figure 4A, has-miR-4779 is predicted to bind to the 3′-UTR of PLK1 mRNA at position 27–33. MG63/Dox cells were transfected with miR-4779 mimic for 48 h, and western blotting analysis showed that PLK1 expression was significantly decreased in MG63/Dox cells (Fig. 4B). Conversely, MG63/Dox cells were transfected with miR-4779 inhibitor for 48 h, and the western blotting analysis showed that PLK1 level was significantly increased in MG63/Dox cells. These results demonstrate that has-miR-4779 negatively regulates the expression of PLK1 in MG63/Dox cells.

Has-miR-4779 regulates the expression of PLK1.
Transfection of miR-4779 mimic or miR-4779 inhibitor affects cell viability of MG63/Dox cells
MiR-4779 mimic and miR-4779 inhibitor are chemically modified, double-stranded miRNA-like RNAs. Both were used to test the function of mature endogenous miRNA upon transfection. To identify the role of miR-4779 in Dox-resistant MG63 cells, miR-4779 mimic or mimic-negative control was used to transfect MG63/Dox cells for 24, 48, and 72 h. Cell viability was determined by MTT assay, and the results showed that OD490 values of MiR-4779 mimic-transfected MG63/Dox cells were significantly lower than those of transfected mimic-negative control, and the decrease varied in a time-dependent manner (**p < 0.01). Conversely, MTT assay results showed that transfection of MG63/Dox cells with MiR-4779 inhibitor resulted in significantly higher cell proliferation than that by transfection with the negative control inhibitor (**p < 0.01) in both 48 and 72 h groups. Moreover, PLK1 vector-transfected MG63/Dox cells were transfected with MiR-4779 mimics for different periods of time. The MTT assay results showed that cell proliferation was not obviously changed compared with that of untreated cells (p > 0.05). Conversely, transfection with miR-4779 inhibitor increased the cell proliferation of MG63/Dox cells; however, cotransfection of PLK1 siRNA clearly resulted in higher inhibition of MG63/Dox cell viability than that in inhibitor control transfected cells (**p < 0.01, ##
p < 0.01). As shown in Figure 5, PLK1 expression appears to promote cell proliferation of drug-resistant
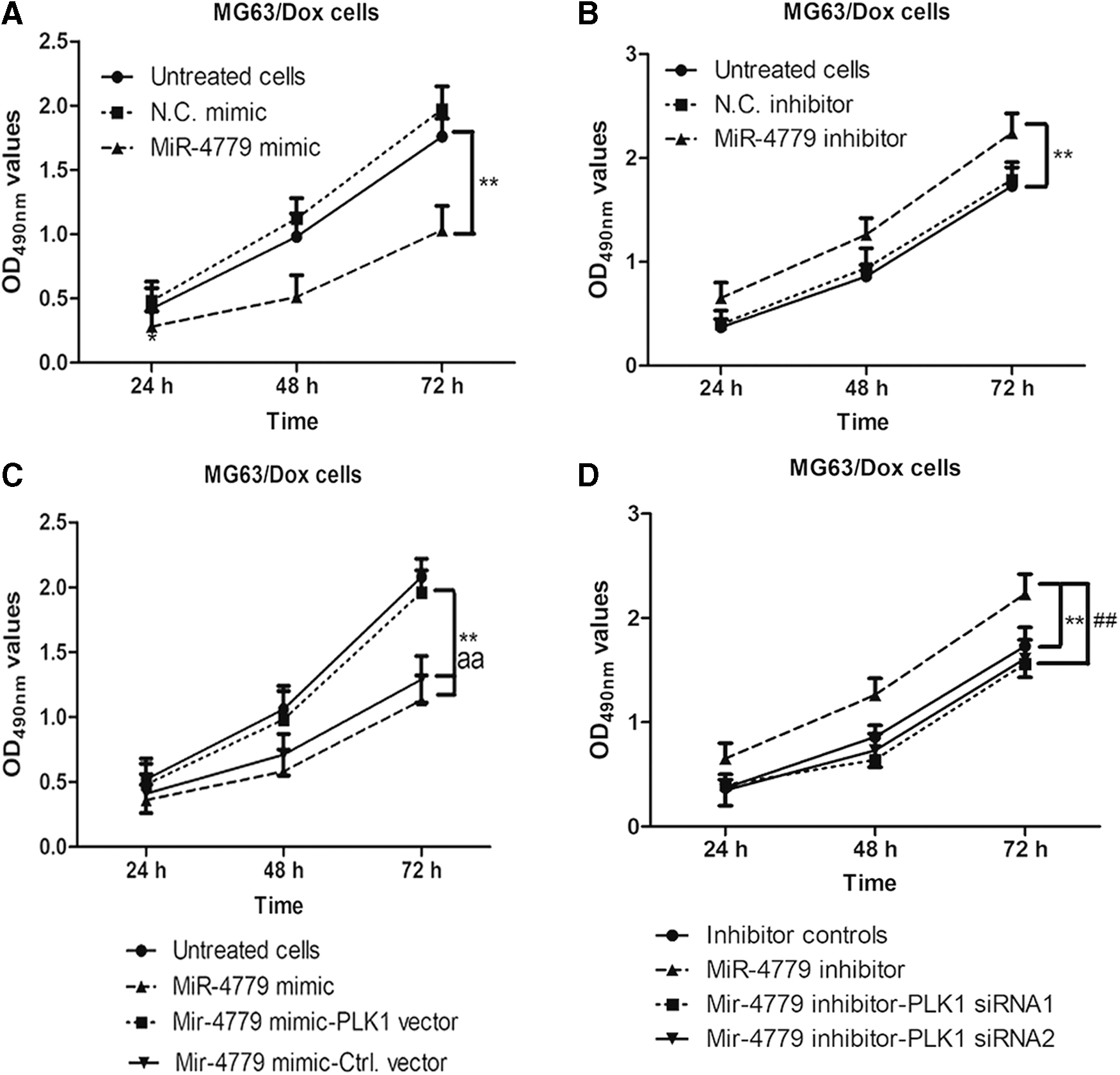
Transfection of miR-4779 mimics or miR-4779 inhibitor affects cell viability of MG63/Dox cells.
Discussion
In the present study, we explored the role of PLK1 in drug-resistant
MiRNAs are a class of short, non-coding RNAs that bind to the 3′-UTR mRNAs of the target genes to negatively regulate gene expression posttranscriptionally. In the present study, we predicted the possible miRNA targeted to the 3′-UTR of PLK1 mRNA using TargetScan software. The results showed that has-miR-4779 binds to the 3′-UTR of PLK1 mRNAs at positions 27–33. Moreover, western blotting data revealed that miR-4779 negatively regulates the expression of PLK1 in MG63/Dox cells. Furthermore, miR-4779 mimic/inhibitor or mimic/inhibitor-negative controls were transfected into MG63/Dox cells for 24, 48, and 72 h. Cell viability was determined by MTT assay, and the results showed that miR-4779 mimics suppressed the MG63/Dox cells, while miR-4779 inhibitor significantly promoted higher cell proliferation of MG63/Dox cells than that in the negative control group. Thus, we concluded that miR-4779 negatively regulates PLK1 expression in MG63/Dox cells. At the same time, overexpression of miR-4779 or interference with the expression of PLK1 inhibits cell proliferation of drug-resistant OS cells.
Duan et al. (2010) optimized a systematic screen of known kinases in OS cell lines and confirmed the effectiveness of PLK1 as a potential drug target for
Because it was inconvenient to collect clinical samples, we have not previously checked the expression of PLK1 and MiR-4779 in clinical samples. Measuring these expression levels is a meaningful way to test the relationship of PLK1 and MiR-4779 in patients with
Cell autophagy is a highly conserved biological process, but its role in promoting chemosensitivity or chemoresistance has not been described in OS. In the near future, we plan to explore how miR-4779 regulates autophagy in the proliferation of MG63/Dox cells and how cell autophagy regulates chemosensitivity in the proliferation of OS cells.
In conclusion, we found that miR-4779 negatively regulates the expression of PLK1 and possesses a cancer-inhibiting role, while PLK1 works as an oncogene in drug-resistant
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received.