
Abstract
The Qing-Tibet Plateau is characterized by low oxygen pressure, which is an important biomedical and ecological stressor. However, the variation in gene expression during periods of stay on the plateau has not been well studied. We recruited eight volunteers to stay on the plateau for 3, 7, and 30 days. Human Clariom D arrays were used to measure transcriptome changes in the mRNA expression profiles in these volunteers' blood. Analysis of variance (ANOVA) indicated that 699 genes were significantly differentially expressed in response to entering the plateau during hypoxic exposure. The genes with changes in transcript abundance were involved in the terms phosphoprotein, acetylation, protein binding, and protein transport. Furthermore, numerous genes involved in hematopoietic functions, including erythropoiesis and immunoregulation, were differentially expressed in response to hypoxia. This phenomenon may be one of reasons why the majority of people entering the plateau do not have excessive erythrocyte proliferation and are susceptible to infection.
Introduction
Acute mountain sickness (AMS) is the most common disease caused by reduced oxygen amounts and decreased pressure on a plateau, and severe cases can progress to high-altitude cerebral edema, which is potentially fatal (Hackett and Roach, 2001). Hypoxia is a critical factor for AMS; it initiates a pathophysiological process and causes the symptoms of AMS. When people rapidly enter a plateau, a series of physiological markers change in the body, including increased red blood cell (RBC) counts, hematocrit (Hct), hemoglobin (HGB), and lung ventilation in the respiratory system and decreased arterial oxygen saturation (SaO2) in the blood (Siques et al., 2009).
A previous study demonstrated that several genes, including HIF-1 and HIF-2, could be induced under exposure to a hypoxic environment (Bigham and Lee, 2014). Many of these genes, such as erythropoietin (EPO), vascular endothelial growth factor (VEGF), tyrosine hydroxylase, and endothelin 1 gene (EDN-1), observed in response to acute hypoxia (Sarkar et al., 2003), are associated with physiological changes. In addition, chronic intermittent hypoxia can change the bone marrow microenvironment, resulting in hematopoiesis (Alvarez-Martins et al., 2016). Our previous study also revealed that HGB could increase to more than 170 g/L after rapid entry to the plateau for 3–7 days (acute hypoxia), but the HGB concentration could remain at ∼170 g/L even after a 30-day (chronic hypoxia) stay on the plateau (Li et al., 2018). Thus, there must be a mechanism to regulate excessive erythropoiesis.
Furthermore, a prolonged stay at high altitudes has physiological effects on the immune system. Environmental factors at high altitudes, such as UV exposure, cold, and hypoxia, can affect the immune system, making it more susceptible to cancer, infections, and autoimmune diseases (Mishra and Ganju, 2010). However, little research has been done on the relationship between the effects of high-altitude hypoxia exposure and human immune system, because the detection of immune changes after exposure to high altitude is more complicated in humans than in experimental animals, and the immune status of the population is extremely heterogeneous.
Herein, comparison of transcriptome differences in samples from individuals with or without exposure to hypoxia revealed distinct differences in gene regulation changes indicated regulation of hematopoiesis at the genome-wide level. In this study, we used mRNA GeneChip arrays to obtain peripheral white cell transcriptional information to assess the gene expression landscape in response to acute and chronic hypoxia exposure, and used this information to explore the possible molecular changes involved in hematopoiesis.
Materials and Methods
Study oversight
This study was approved by the Institutional Review Board of the General Hospital of the Air Force, PLA (no. 2017-05-YJ01), and all volunteers provided signed written informed consent. People with lung, heart, and blood diseases were excluded, and eight individuals were recruited to provide samples for RNA sequencing. All were healthy young men who had never previously been at high altitude.
Study procedure
The set of volunteers for the expression profile analyses was assembled at a low-altitude starting point (500 m) and then made a rapid ascent to a high altitude (5200 m) by car over a 72-h period. Furthermore, these people were transported to an altitude of 3800 m. Upon the volunteers' arrival at high altitude, whole blood samples were collected at the starting point and at 3, 7, and 30 days after entry to the plateau. During the exposure to high altitude, all volunteers participated in a regimented daily life and avoided any exercise or physical labor. The volunteers before and after entering the plateau are the same population. All blood samples for mRNA measurements were stored at −80°C with TRIzol in RNase-free tubes for subsequent analysis.
Microarray analysis
Blood samples were collected in BD K2-EDTA tubes. Total RNA was extracted from peripheral white cells with a miRNeasy mini kit following the manufacturer's protocol (Qiagen GmbH, Hilden, Germany). A total of 500 ng of total RNA were used for sample preparation starting from 5 mL of blood. All samples were hybridized on a Human Clariom D (Affymetrix, Santa Clara, CA) gene chip and were analyzed using Transcriptome Analysis Console 4.0 software (Thermo Fisher Scientific, Waltham, MA). The Signal Space Transformation-Robust Multiarray Average (RMA) algorithm was applied for background adjustment, normalization, and log transformation of the signal intensity. Microarray data were deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE135109.
Gene chip data processing and analysis
The Affymetrix Gene chip arrays were first inspected using multiple quality control steps. All arrays were imaged, and no obvious scratches or areas of spatial variation were observed. Digestion curves describing trends in RNA degradation between the 5′ end and the 3′ end of each probe set were examined, showing a notable outlying trend in the same outlying array. To determine the gene expression difference between the three simulation periods, samples taken by volunteers while they were in the plain were used as control group and analysis of variance (ANOVA) was performed on the RMA expression values. Genes identified with an adjusted p value of <0.05 were extracted for further inspection and analysis. Gene annotation was gathered from the Affymetrix NetAffx Analysis Center. Genes were categorized by their biological functions with DAVID Bioinformation Resources (Kalikstad et al., 2017), and p-values were corrected by the Benjamini-Hochberg FDR method. These categories were analyzed using a series of Fisher's exact tests to determine the representation of functional categories by the selected genes.
Confirmatory quantitative real-time PCR
We selected seven genes showing significant differential expression after simulated hypoxic exposure to verify the results of the microarray experiment. RPL13A (NM_012423) was selected as our endogenous control gene (Xiao et al., 2017). The primer pairs for each of these eight genes were selected from PrimerBank (Table 1). Before the quantitative real-time (qRT) PCR experiment, the efficiency of each primer pair was assessed. The source RNA used for the qRT-PCR experiment was derived from another five volunteers. Then, 500 ng of total RNA from these 20 samples representing the three hypoxia treatments were reverse transcribed to cDNA using ReverTra Ace® qPCR RT Master Mix with gDNA Remover (TOYOBO, Osaka, Japan). For each gene, qRT-PCR was performed in duplicate on each cDNA sample. Amplification and data analysis were performed using a CFX96 thermocycler PCR system (Bio-Rad, Hercules, CA) with 40 amplification cycles. Pearson's correlation was used to assess the relationship between the qRT-PCR data and microarray data.
Primer Sequence Information for Quantitative Real-Time PCR Amplification Used in This Study
Results
Physiological response to hypoxia
After exposure to hypoxia for 3, 7, and 30 days, the RBC counts, HGB, and Hct differed significantly (p < 0.001) among the three periods and compared to those in normoxic subjects. People on the plateau for 3 days had a significant increase in the mean RBC count (5.5 × 1012/L), HGB (171 g/L), and Hct (52.7%), which could last for seven or even 30 days (Fig. 1).

Mean RBC counts, HGB, and Hct of individuals after exposure to a hypoxic environment. Whole blood was collected from the volunteers after entering the plateau. RBC counts
Identification of differentially expressed genes
Peripheral white cells were obtained from volunteers with the indicated hypoxic exposure times, while peripheral white cells from subjects without hypoxic exposure were used as controls. Human Clariom D arrays were used for transcriptomic analyses of peripheral white cells. According to the manufacturer's algorithm, a total of 699 genes (Fig. 2) with at least a 1.5-fold change in expression and a p value of <0.05 were identified. These genes included 386 downregulated genes and 313 upregulated genes (Supplementary Table S1).

Overview of mRNA expression. The Venn diagram indicates the number of transcripts equally and differentially expressed in peripheral white cells derived from individuals after entering the plateau.
Functional annotation of differentially expressed genes
To explore the similarities between the transcriptomes of these subjects, cluster analysis was performed on the differentially expressed genes (DEGs) (Fig. 3). Although there were variations in expression within the replicates of individual groups, the heat map representation of the gene expression patterns revealed a distinct trend of selected differentially expressed genes during different exposure times.
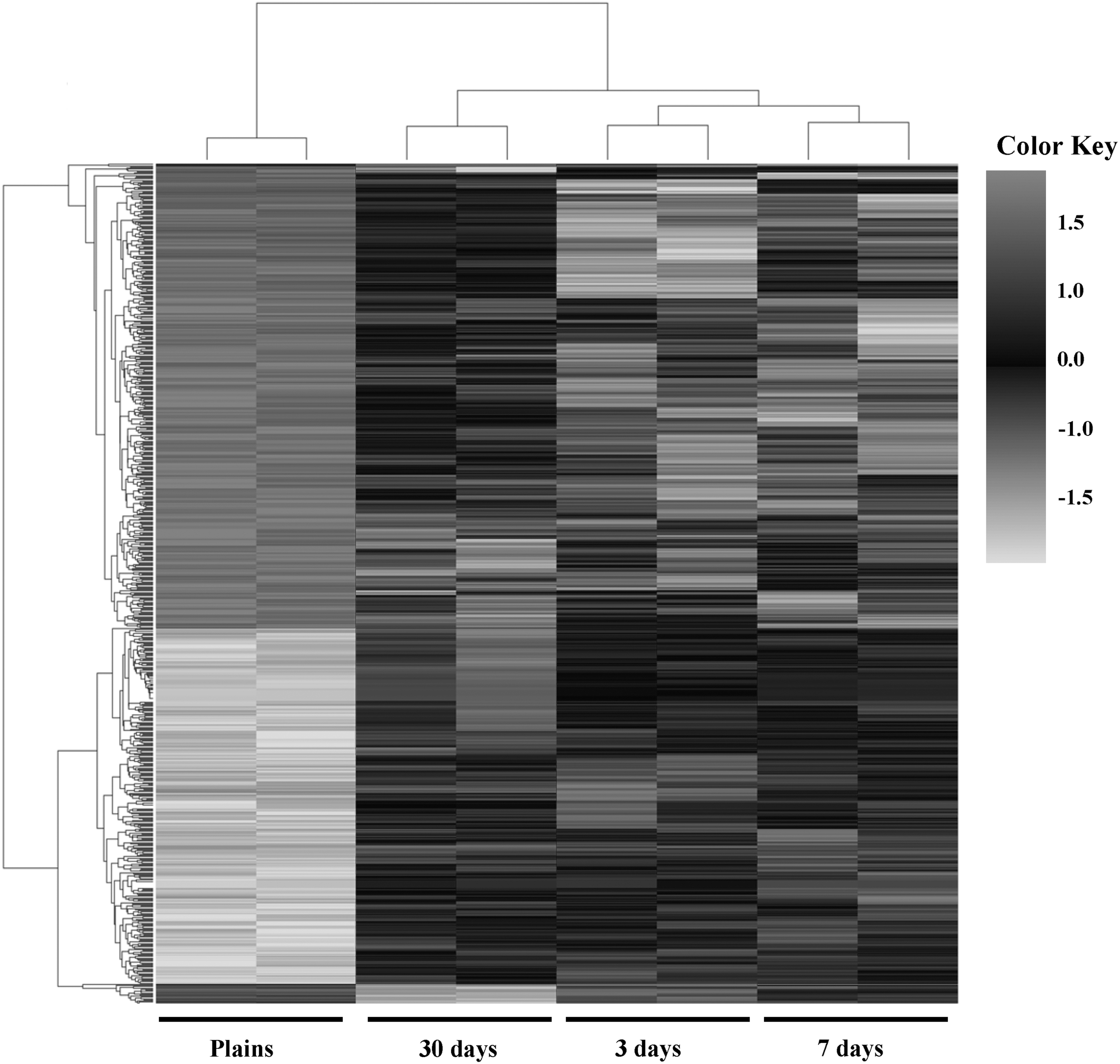
Hierarchical heat map of differential gene expression as determined by Human Clariom D array analysis in peripheral white cells. Hierarchical cluster analyses of the differentially expressed genes (p < 0.05) in individuals staying on the plateau for 3, 7, or 30 days compared to individuals on the plain without hypoxic exposure. The dark gray area indicates highly upregulated genes, and the light gray area indicates downregulated genes.
To determine the biological significance of the DEGs, we performed functional annotation of these genes with DAVID Bioinformation Resources. The genes were mainly enriched in the terms phosphoprotein, acetylation, protein binding, and protein transport (Table 2). Furthermore, 10 significant Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Fig. 4) at p < 0.05, such as cell cycle, T cell receptor signaling pathway, viral carcinogenesis, protein processing in endoplasmic reticulum, and metabolic pathways, were enriched.

KEGG analysis of DEGs in individuals after hypoxic exposure. The DEGs were analyzed using the online tool DAVID and the molecular pathway database KEGG. A number of signaling pathway annotations that were significantly enriched were identified. DEG, differentially expressed gene; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Functional Annotation of Different Expressed Genes After Entering into High Altitude by DAVID Analysis
Confirmatory qRT-PCR
As mentioned earlier, our aim was to explore the changes in hematopoietic regulation in hypoxic environments. Interestingly, we found seven genes (CXCR3, JAK1, SLAMF6, NF-κB1, NF-κBIA, NF-κBIZ, and NF-κBIE) that participated in hematopoiesis (Garcia-Lopez et al., 2001; Li et al., 2014; Wang et al., 2016). To validate the expression profiles determined by the Human Clariom D expression array analysis, qRT-PCR was performed on these genes (Fig. 5) in samples from another five individuals. We performed linear regression analysis to evaluate the relationship between the microarray data and PCR data, which resulted in a very strong correlation of 0.99. This result indicated a high correlation and confirmed our microarray results (Table 3).
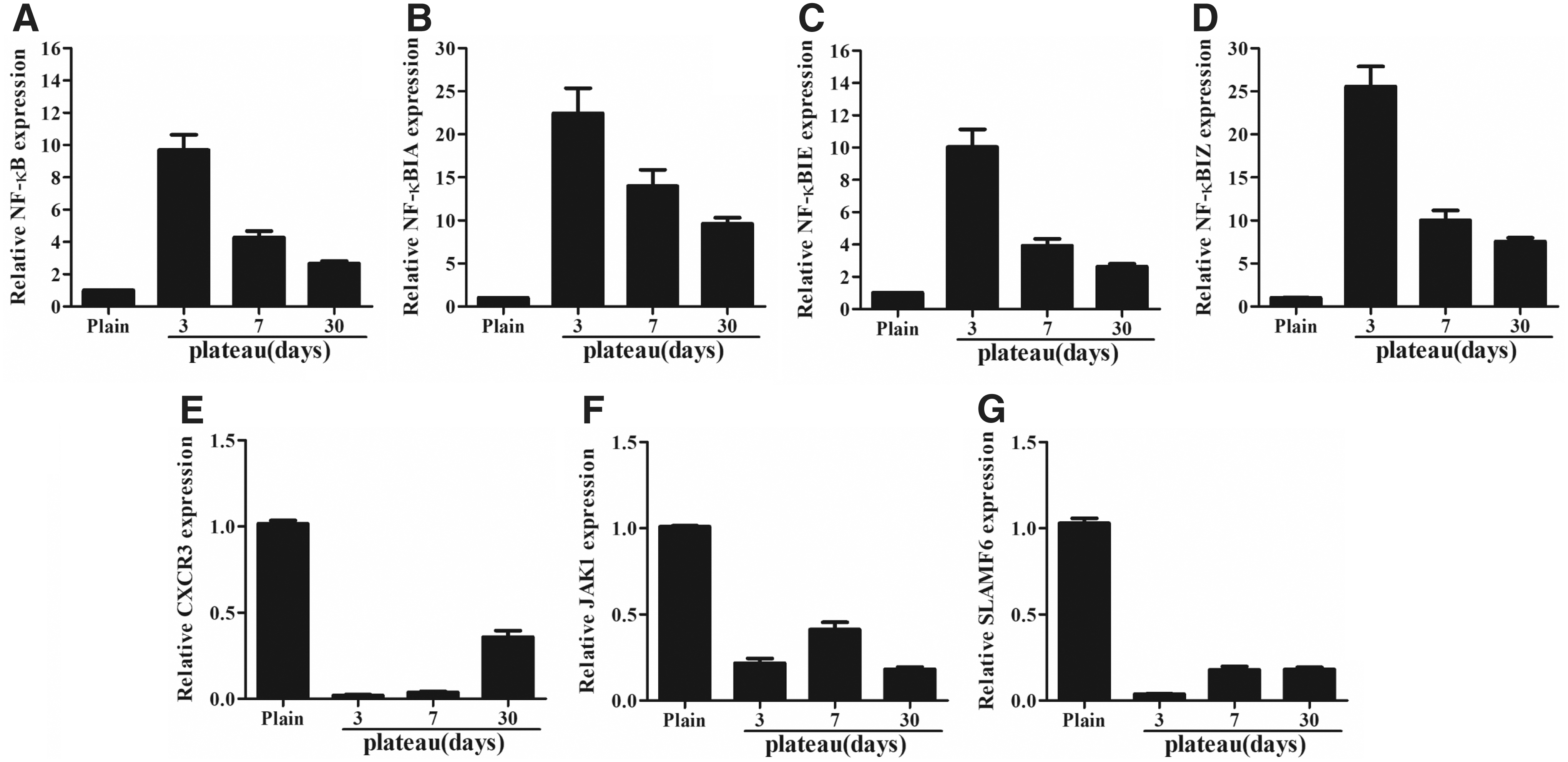
Confirmatory qRT-PCR of seven transcripts. Comparison between the expression values from the microarray analysis and expression values from qRT-PCR. NF-κB, nuclear factor kappa B
Correlation of Seven Transcripts Between Quantitative Real-Time PCR and Microarray Analysis
Discussion
Hypoxia is a major environmental condition that impacts the regulation of expression of several hundred genes involved in acclimatization to a limited oxygen supply. In this study, we performed RNA microarray analyses in white blood cells isolated from whole blood cells of individuals who stayed on the plateau for different durations to explore the changes in gene expression with time under hypoxic exposure.
Based on the obtained difference in RBC counts, HGB, and Hct among different individuals, the hypoxic environment was effective in eliciting a significant physiological response. A previous study indicated that Endothelial PAS domain-containing protein 1 (EPAS1) activates the transcription of its downstream target gene EPO under hypoxic conditions (Rankin et al., 2007); thus, the increase in RBCs or HGB could improve the oxygen supply to tissues, allowing individuals to acclimate to the hypoxic conditions (Peng et al., 2017). In a hypoxic environment, EPAS1 may accumulate in the cytoplasm and then translocate into the nucleus and bind to the hypoxia response element to activate the downstream expression of EPO (Petousi and Robbins, 2014).
However, few genes regulated by hypoxia-inducible factor (HIF) were included among the 699 genes significantly differentially expressed during the hypoxic exposure period. This result indicated that after more than 3 days of exposure, the previously described HIF pathway was no longer a significant factor in regulating and maintaining physiological acclimation to hypoxia. Indeed, studies have shown that the HIF abundance peaks after 5 h of hypoxic exposure and then returns to normal after a few hours (Stroka et al., 2001). Furthermore, our previous study also showed that HIF-regulated EPO exhibited a consistent expression pattern (Li et al., 2018). These findings indicated that even though HIF is a putative transcription factor in the regulation of acute hypoxia, it is not important in maintaining acclimatization to chronic hypoxic stress. The reason for this phenomenon may be that tissues return to oxygen homeostasis, and various stress responses and HIF pathways are no longer activated. Hypoxic exposure enters a chronic state, and other pathways or factors are activated, which regulate processes by maintaining the acclimatization state.
Mild increases in HGB are generally seen as beneficial for acclimation to plateau altitudes (Bozzini et al., 2005). However, the most dramatic result of this study was the rapid increase in HGB and erythrocytes after a 3-day stay on the plateau. This increase indicated that the expression of hypoxia-related genes and erythropoiesis-related genes should increase during hypoxic exposure. For example, the expression of the hypoxia-related gene, VEGF, was significantly upregulated during high altitude exposure for 3–7 days, but then returned to normal levels after 30 days of acclimation (Supplementary Table S1). However, according to the gene chip and qRT-PCR results, the erythropoiesis-related NF-κB inhibitor (α, ɛ, and θ) genes were upregulated after exposure to a hypoxic environment, resulting in NF-κB/Wnt pathway inhibition (Li et al., 2014; Ma and Hottiger, 2016) and subsequent blockade of erythropoiesis. This finding may explain why HGB and erythrocytes do not overproliferate in people rapidly entering high altitudes.
Moreover, hypoxic exposure not only affects erythropoiesis but also affects other systems, such as the immune system. Epidemiological data show that residents at high altitudes are more susceptible to pneumonia than those at low altitudes, and the incidence and mortality of pneumonia are greatly increased among residents at high altitude, suggesting that hypoxia may damage the body's defense functions and increase its susceptibility to disease (Singh et al., 1977).
Further studies have shown that high-altitude hypoxia can induce immunosuppression (Mishra and Ganju, 2010; Oliver et al., 2013; Morabito et al., 2016), but the mechanism is still unclear. In this study, several immune-related genes, such as CXCR3 and JAK1, were downregulated during exposure to the hypoxic environment. CXCR3 is a G protein-coupled, seven-subunit, membrane-penetrating receptor that is expressed in a variety of inflammatory cells, such as vascular endothelial cells, activated lymphocytes, macrophages, and dendritic cells (Garcia-Lopez et al., 2001), but not in T lymphocytes, B lymphocytes, and granulocytes during the resting period. CXCR3 and CCR5 can be expressed in Th1 cells, and CXCR3 can be used as a marker of activation of Th1 lymphocytes (Bonecchi et al., 1998). The ligand of CXCR3, interferon-gamma-inducible protein (IP-10/CXCL10), can be closely connected with CXCR3, which participates in the activation, migration, and infiltration of T lymphocytes (Colvin et al., 2004). In addition, JAK1, a molecule associated with thymus development and maturation, was also significantly downregulated under high-altitude hypoxia. These results indicated that hypoxic exposure could indeed participate in immune suppression. However, the SLAMF6 gene, as an inhibitory receptor that controls the production of IFN-γ-producing CD4+ cells and Tfh cells (Wang et al., 2016), was significantly downregulated after hypoxic exposure, which indicated that downregulation of SLAMF6 can enhance immune function after entry to a hypoxic environment. The reason for this contradiction may be that the body's immunoregulatory function could not be oversuppressed or overenhanced in the hypoxic environment, but was maintained in a homeostatic immune state.
Conclusion
The detection of changes in hematopoiesis after exposure to the plateau environment is more complicated in humans than in experimental animals. In this study, we observed striking differential gene expression pattern related to hematopoiesis after entry to the plateau. The gene expression profile showed some important differences in erythropoiesis-related and immune-related genes, revealing why the majority of people entering the plateau do not have excessive erythrocyte proliferation and are susceptible to infection. The work performed in this study may provide a typical demonstration of approaches for deciphering the mechanism of high-altitude adaptation in the future.
Footnotes
Acknowledgment
The authors thank Ying Wang and Guoxing You for helpful discussion and review of this article.
Ethical Approval
This study was approved by the Institutional Review Board of the Air Force Medical Center, PLA (2017-05-YJ01), and the research was carried out in accordance with the World Medical Association Declaration of Helsinki. All volunteers signed informed consent forms, and personal background investigations were conducted to rule out kinship among them.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Military Major Special Project of PLA, China (no. AWS13J004), and Key Logistics Research Projects of PLA, China (no. BWS16J006-03-01).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.