
Abstract
Kearns–Sayre Syndrome (KSS) is a severe mitochondrial disorder involving the central nervous system, eyes, ears, skeletal muscles, and heart. The mitochondrial DNA (mtDNA) rearrangements, especially the deletions, are present in almost all KSS patients and considered as the disease-causing factor. However, the size and position of mtDNA deletions are distinct in different individuals. In this study, we report the case of a pair of Chinese twins with KSS. The twin patients revealed typical KSS clinical symptoms, including heart block, bilateral sensorineural hearing loss, progressive external ophthalmoplegia, exercise intolerance, proximal limb weakness, and endocrine disorders. Using long-range polymerase chain reactions (long-range PCR) and next-generation sequencing (NGS), the genetic features of the twin patients were investigated. A large 6600 bp mtDNA deletion, ranging from position 8702 to 15,302, was detected in both patients. To our knowledge, this kind of mtDNA deletion has never been described previously. Our study enriched the mutation spectrum of KSS and showed that NGS is a powerful tool for detecting mtDNA large variants.
Introduction
Kearns–
The spontaneous mitochondrial DNA (mtDNA) rearrangement, including deletion and duplication, is identified to be the main cause of KSS (Moraes et al., 1989; Chinnery et al., 2004; Dominic et al., 2014). The mitochondria are special organelles in cells, which contain their own DNA (mtDNA) and independent replication, transcription, and translation systems. They carry out various tasks, including pyruvate oxidation, the Krebs cycle, the metabolism of amino acids, fatty acids, steroids, and adenosine triphosphate (ATP) generation in cells (Raha and Robinson, 2000; Chan, 2006). Actually, there are only 37 genes in human mtDNA: 13 genes encoded the subunits of the respiratory chain and other genes are needed for mtDNA translation (DiMauro and Schon, 2003; Kokotas et al., 2007). The mutations on the mtDNA lead to disruption of the respiratory chain function of many tissues, especially in those with a high-energy demand, such as muscles and the brain, and cause serious pathological symptoms, including KSS, Pearson's syndrome (PS), PEO, aminoglycoside-induced deafness, and so on (DiMauro and Schon, 2003; Chinnery et al., 2004; Alemi et al., 2007; Yan et al., 2011; Bannwarth et al., 2013).
In the last few decades, a variety of mtDNA deletions, duplications, and point mutations encompassing subunits of the oxidative phosphorylation enzymes and t-RNA genes are found in most cases of KSS disease (Moraes et al., 1989; Zeviani et al., 1989; Bosbach et al., 2003; Puoti et al., 2003). The mtDNA deletions vary in location and size. In 1989, Schon et al. found large-scale mtDNA deletions in KSS patients with the size ranging from 1.3 to 7.6 kb (Moraes et al., 1989). Among these mutations, a 4.9 kb “common deletion” was identified as it was present in different patients and two 13-bp mtDNA repeats (positions 8470 to 8483 and 13,447 to 13,460) were predicted as hotspots for this deletion. Subsequently, another 7436-bp common deletion flanked by a 12-bp direct repeat (8637–8648 and 16,073–16,084) was found in KSS patients (Wei et al., 1996).
Moreover, several uncommon mtDNA deletions were also reported. In 1995, a 3.1 kb mtDNA deletion (11,259–14,368) was found in the muscle of a German patient with KSS (Klopstock et al., 1995). Sokolova et al. (2002) identified a heteroplasmic 5.5 kb deletion, encompassing nucleotides 8278–13,770 of the mtDNA, in a KSS patient from a Russian family. Moreover, two novel mtDNA deletions, the 2.3 kb deletion (12,113–14,421) and the 7.7 kb deletion (6330–13,993), were found in KSS patients (Lertrit et al., 1999). Two uncommon mtDNA 9.768 kb and 7.253 kb heteroplasmic deletions at the position of 6124–15,893 and 8572–15,826 were also detected in blood leukocytes, buccal mucosa, and hair follicles of KSS patients (Mkaouar-Rebai et al., 2010). However, analysis of additional populations is still important to broaden the mutational spectrum of KSS disease. Further understanding the mechanism of the mtDNA deletion and KSS disease might be beneficial for developing the treatment of KSS as well as other mitochondrial disorders.
Materials and Methods
Our institutional approval number for human studies is 2,017,044.
Patients
The probands were a pair of twin boys, born from a nonconsanguineous family. There was no family history of mitochondrial disorders, diabetes, hypertension, trauma, bacterial meningitis, or cardiac disease. The parents of the twins were healthy and lived in a small city in Province Xinjiang, China (Fig. 1A). The pregnancy and delivery were normal. Their living conditions were quite well, and they had no bad personal habits or hobbies. The twins had the same disease history and symptoms. They had normal growth and development during early childhood. However, they begin to show signs of short stature and underweight and developed bilateral eyelid ptosis at the age of 7 years, followed by PEO and PR. The twins visited EENT Hospital of Fudan University because of hearing loss at the age of 11 years. All family members were recruited and informed about the scope and requirements of the study and signed the patient consent form approved by the Ethics Committee of Fudan University.

Pedigree of the twin patients and the phenotype of progressive external ophthalmoplegia.
Clinical Evaluation
After obtaining informed consent from all the family members, a set of clinical examinations were carried out, including neurological, cardiological, audiological, as well as endocrine examinations. Briefly, the audiological examinations consisted of otoscopy, puretone audiometry (PTA), immittance, distortion product otoacoustic emission, and auditory brainstem response. The PTA data were presented as average value of audiometric thresholds at 250, 500, 1000, 2000, and 4000 Hz. Normal hearing was classified as a PTA ≥20 dB HL, mild hearing loss as a PTA ≥20 dB HL and ≤40 dB HL, moderate hearing loss as a PTA ≥41 dB HL and ≤70 dB HL, severe hearing loss as a PTA ≥70 dB HL and ≤95 dB HL, and profound hearing loss as a PTA ≥96 dB HL. Since the kids revealed short stature and stunting, endocrine work-up was performed to identify endocrinopathy, including the examinations of adrenocorticotropic hormone (ACTH), parathyroid hormone (PTH), thyroxin, growth hormone (GH), cortisol, prolactin (PRL), progesterone, follicle-stimulating hormone (FSH), testosterone, luteotropic hormone (LH), estradiol, adenosine deaminase (ADA), serum phosphate, serum calcium, as well as CSF analyses. Furthermore, the neurological examination, brain magnetic resonance imaging (MRI), and computer tomography (CT) scanning were performed to check the abnormalities of brain. Electrocardiogram (ECG) detection was used to record to rule out the cardiological problems.
DNA extraction
After obtaining the consents from all the family members, 5 mL peripheral blood was withdrawn for the extracting of mtDNA. The Qproteome Mitochondria Isolation Kit (Qiagen, Hilden, Germany) was used to purify the mitochondria from white blood cells. The mtDNA was extracted using the QIAamp DNA Mini Kit (Qiagen).
Identification of deletion breakpoints by long-range PCR
Long-range polymerase chain reaction (PCR) was employed to detect the KSS-associated deletion in the twins using 2 × G5 High-Fidelity PCR Mix (NovaBio). The following primers were used: 5′-CCGCACAAGAGTGCTACTCTCCTC-3′and 5′-GATATTGATTTCACGGAGGATGGTG-3′. The PCR amplification was performed using the long-range PCR enzyme mix. The conditions for the PCR reaction were: 95°C, 3 min; 30 cycles of 95°C, 15 s; 60°C 15 min; and then 72°C for 8 min. Products were separated on 0.8% agarose gel and visualized with ethidium bromide.
NGS for the mitochondrial genome and data analysis
mtDNA (5 μg) was fragmented ultrasonically with the Covaris E210 DNA shearing instrument (Covaris, Inc., Woburn, MA) to an average size of 300 bps for subsequent construction of Illumina NGS libraries. The Covaris protocol is set at 3 min total duration, duty cycle 10%, intensity 5, 1 and 200 cycles per burst. Sequencing libraries were prepared using the NEBNextTM DNA Sample Prep Master Mix set (E6040; NEB Biolabs, Ipswich, MA) following the manufacturer's protocol. NGS was performed on the Illumina Hiseq2000 machine in paired-end 100 bps modes. FASTQ files were produced by an Illumina CA1SAVA v1.8 pipeline and aligned to human reference genome (hg19) using the Burrows-Wheeler Aligner (BWA) program.
In our study, FASTQ data files generated after sequencing with the Illumina HiSeq2000 were processed using the DNAnexus with bioinformatic data processing pipelines (
Results
The probands (II-1, II-2) and their parents (I-1, I-2) were enrolled in our study (Fig. 1). As mentioned in past medical histories, the twin boys developed bilateral eyelid ptosis at the age of 7 years, followed by PEO and PR. There was no family history of hearing loss or KSS disease. Pure-tone and acoustic immittance testings, otoscopic examinations, as well as complete physical examinations were performed in all family members. The PTA tests showed that only the twins had bilateral severe sensorineural hearing loss in a slope form (Fig. 2). As the twins affected with short stature and underweight, they were only 1.3 m in height and 29 kg in weight until 11 years of age. To detect GH deficiency, endocrine work-up was carried out and the results are summarized in Table 1. The twins were diagnosed as hypoparathyroidism for low PTH levels [7.70 pg/mL (II-1), 11.56 pg/mL (II-2), 15–65 pg/mL (normal range)], severe hypocalcemia [1.81 mM (II-1), 1.44 mM (II-2), 2.20–2.65 mM (normal range)], and hyperphosphatemia [2.04 mM (II-1), 2.83 mM (II-2), 0.8–1.6 mM (normal range)]. Further analysis revealed markedly elevated serum ACTH: 195.14 pg/mL in twin II-1 and 77.18 pg/mL in twin II-2 (the normal level is 7.9–32.1 pg/mL). Moreover, the endocrine detecting showed GH deficiency and sex hormone deficiency in the twins: the maximum level of GH for the twin boys was 7.27 ng/mL and 7.01 ng/mL, which may account for the short stature and stunting. Their liver function, amylase, lipase, and blood cell count were normal, antinuclear, and anti-DNA antibodies were negative based on these evidences. The twins also suffered from cardiac conduction defects. The standard ECG examinations revealed a right bundle branch block in both twin boys (Fig. 3). Brain MRI demonstrated bilateral and symmetrical signal changes in bulbus medullae, cerebral ganglia, and mesencephalon (Fig. 4). Based on both the clinical manifestations and accessory findings a diagnosis of KSS was made.

The PTA was detected in the probands. Frequency in hertz (Hz) is plotted on the x-axis and the auditory threshold in decibels (dB) on the y-axis.
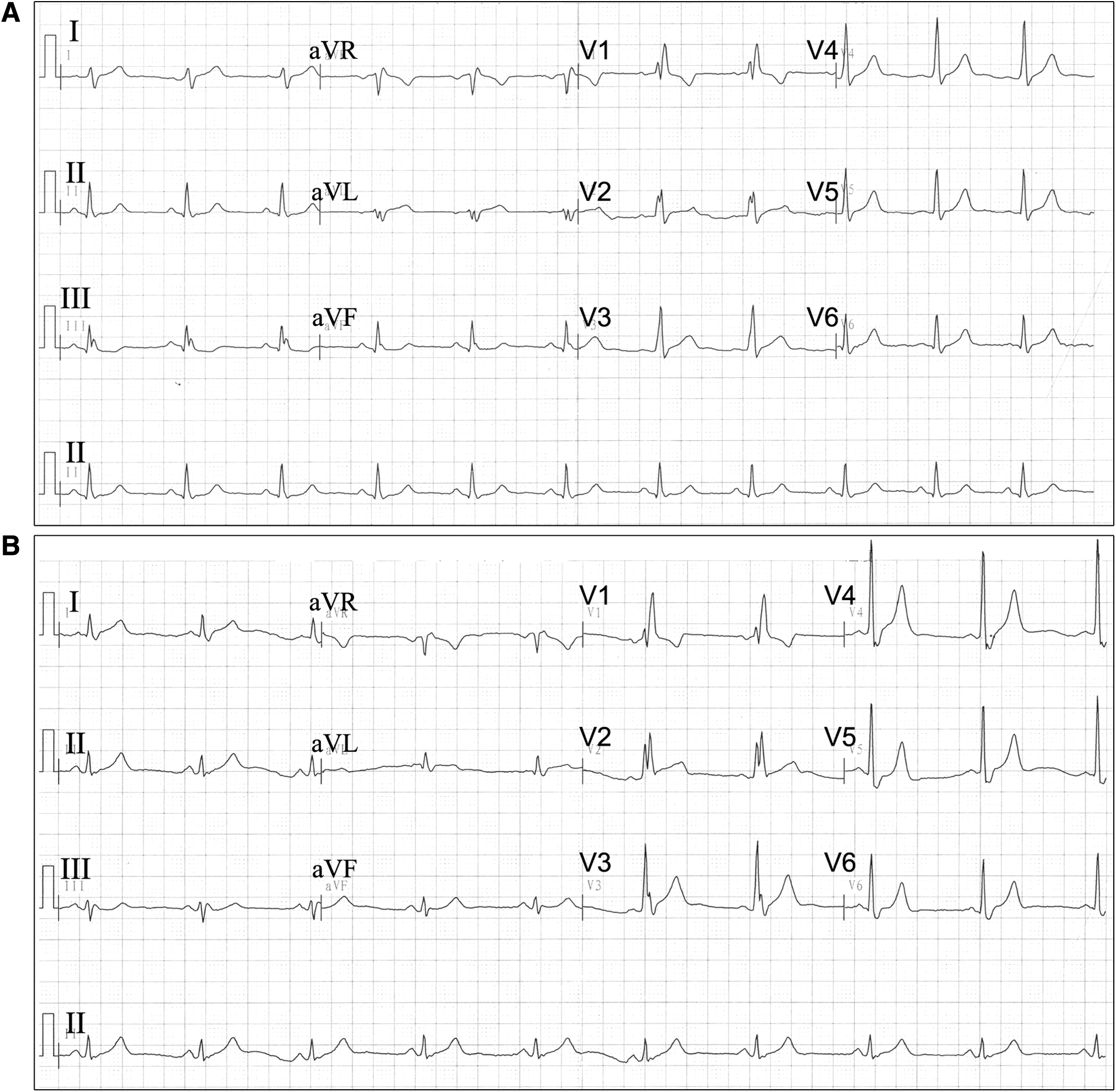
Standard ECG showing RBBB in patients II-1
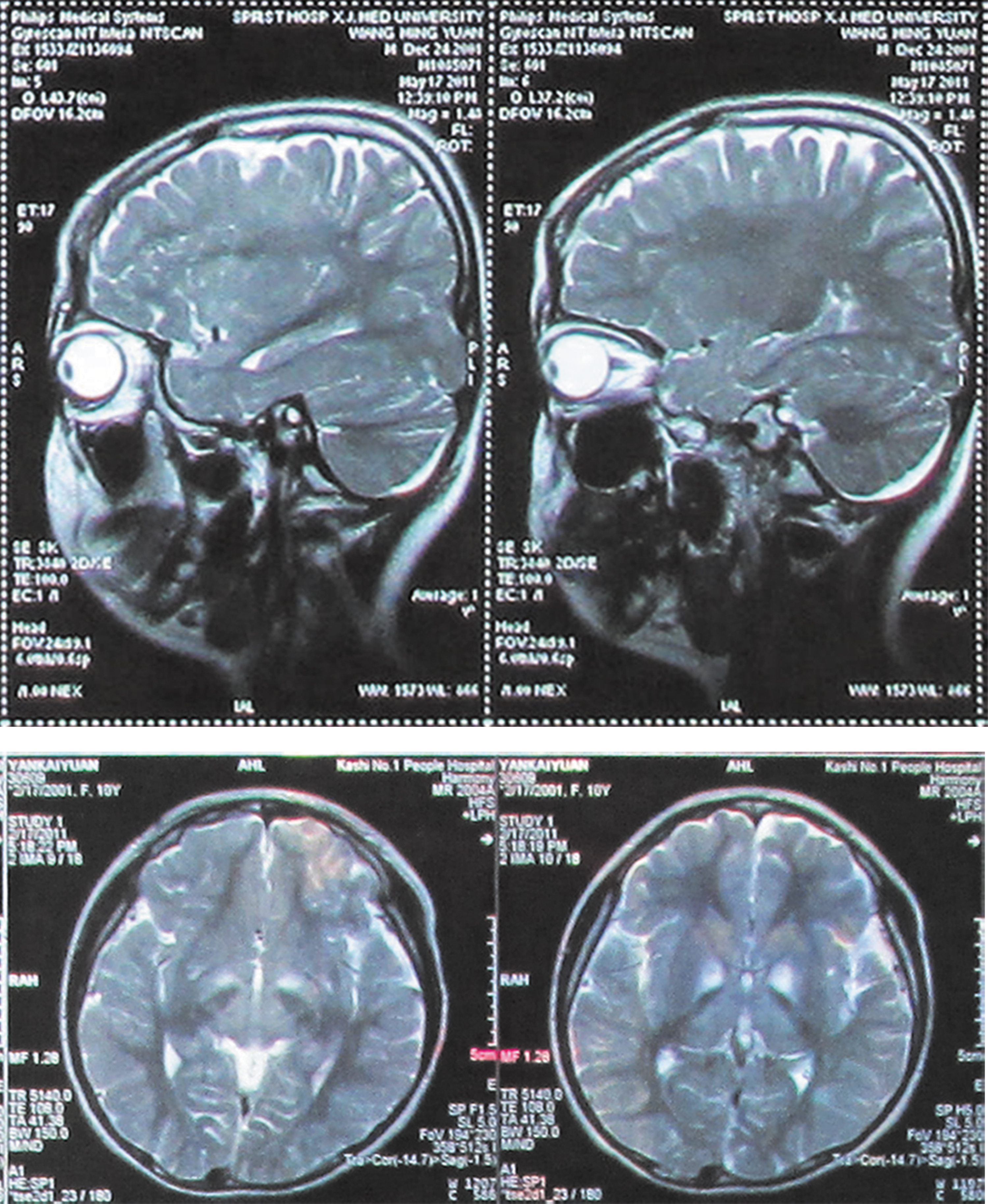
MRI of the patient's brain showing bilateral and symmetrical signal changes in bulbus medullae, cerebral ganglia, and mesencephalon. MRI, magnetic resonance imaging.
Hormones and Other Biochemical Parameters in Blood Samples
Boldface indicates statistical significance.
Lack of CRH raised the level of ATCH in both twins.
PTH deficiency is considered a main characteristic of the disorder.
Growth hormone (GH) is synthesized and secreted by somatotrophs in the anterior pituitary, which function may be affected by mitochondrial dysfunction.
Increased progesterone in both male twins reflects endocrine dysfunction.
Testosterone, Luteinizing hormone (LH), Estradiol, and ADA are affected by the endocrine disorder.
Parathyroid hormone (PTH) regulates the levels of calcium and phosphorus in blood, the dysfunction of Parathyroid caused by the syndrome.
Twins may have been suffering slight nerve inflammation.
mtDNA deletion is the main cause of KSS. To investigate the possible deletions on mtDNA, long-range PCR was performed on the twin boys. As shown in Figure 5, the long-range PCR yielded a product of ∼17 kb in mother (Lane 3), father (Lane 4), and normal control (Lane 5) and a product of ∼10 kb in both patient II-1 (Lane 1) and II-2 (Lane 2), which indicated a ∼7 kb deletion in the mtDNA of patients. Furthermore, by comparing lane 1 and lane 2, we found the mtDNA of patient II-2 weighted less compared with patient II-1 and it is obvious that the deletion part of II-2 has more coverage than II-1 within the deletion region. It is possible that II-2 had a higher ratio of cells with mtDNA deletion than II-1.
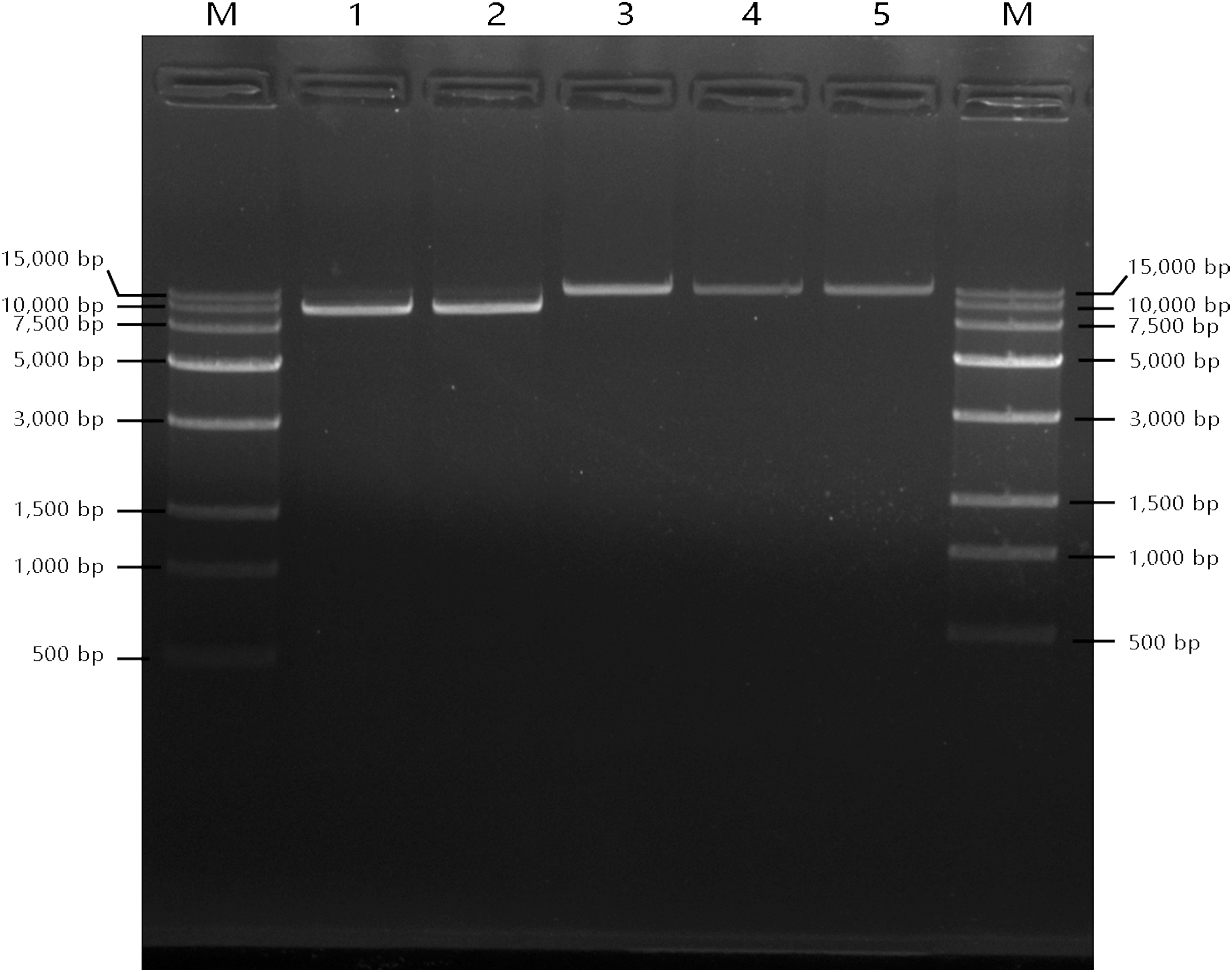
Agarose gel electrophoresis results for long-range PCR amplification of mitochondrial DNA. The different kinds of long-range PCR products were run on 4% agarose gel. All the products were amplified from the mtDNA from white cells in peripheral blood. Line M, 20,000 bps ladder; lane 1, the mtDNA with a large deletion amplified from mtDNA isolated from the white cells in the KSS Twin 1; lane 2, the mtDNA with a large deletion amplified from mtDNA isolated from the white cells in the KSS Twin 2; lane 3, the mtDNA amplified from the healthy mother; lane 4, the mtDNA amplified from the healthy father; Line 5: the mtDNA amplified from the normal control. mtDNA, mitochondrial DNA; KSS, Kearns–Sayre Syndrome.
To confirm and locate the detection of mtDNA deletion in the KSS patients, the genetic analysis of mtDNA was performed by the NGS method. The depth of coverage for the whole mitochondrial genome was presented using DNAnexus. As shown in Figure 3, a 6,600 bp deletion was found in the mtDNA of patient II-1 and II-2, which is consistent with the results of long-range PCR. Next, the deletion located between nucleotides 8, 702 and 15, 302, covering more than one third of the mtDNA (the whole length of 16,571 bps of the sequenced mtDNA). Moreover, the heteroplasmy level for II-1 and II-2 were 78.3% and 93.7%, which confirmed our prediction base on the results of long-range PCR (Fig. 6).

Results of the genetic analysis of patients' mtDNA. Mapping results from DNAnexus for the whole mitochondrial DNA. Arrow A: The whole length of the mitochondrial chromosome; Arrow B: the deletion of twin1; Arrow C: the deletion of twin2.
Discussion
KSS is a multisystem disorder involving the central nervous system, eyes, ears, skeletal muscles, and heart (DiMauro and Schon, 2003; Kokotas et al., 2007; Dominic et al., 2014). Due to the lack of effective therapeutic approaches, KSS is ultimately fatal. As the clinical symptoms progress, the patients usually died before the age of 30 years (Moraes et al., 1989; DiMauro and Schon, 2003). In this case, a pair of twin boys was diagnosed with KSS for the typical clinical symptoms: chronic PEO, PR, sensorineural hearing loss, GH deficiency, hypoparathyroidism, and cardiac conduction defect. Using long-range PCR, we identified a novel 6600 mtDNA deletion in the twins, but not their healthy parents. The genetic analysis base on the NGS data confirmed the common large-scale deletion and located the deletions between 8702 and 15,302 of the mtDNA in the KSS twins.
Mitochondria are cellular organelles involved in calcium signaling, apoptosis, intermediary metabolism, and bioenergetics (Raha and Robinson, 2000; DiMauro and Schon, 2003; Cherry et al., 2016). Different with other cellular organelles, mitochondria had their own DNA and independent protein synthesis system. The so-called mtDNA encoded essential components of the mitochondrial respiratory chain, which drives the synthesis of ATP. Structurally, the functional respiratory chain consists of five enzyme complexes: Complex I, the reduced nicotinamide adenine dinucleotide dehydrogenase/ubiquinone oxidoreductase (∼46 subunits); Complex II, succinate dehydrogenase/ubiquinone oxidoreductase (4 subunits); Complex III, ubiquinone/cytochrome c oxidoreductase (11 subunits); Complex IV, cytochrome c oxidase (13 subunits); and Complex V, ATP synthase (∼16 subunits) (DiMauro and Schon, 2003; Kokotas et al., 2007). Two small electron carriers, ubiquinone (coenzyme Q10) and cytochrome c, are also essential for the respiratory chain. Mitochondria DNA deletions are found to cause mitochondrial disorders, such as KSS, PEO, PS, NARP (Neuropathy, Ataxia, and Retinitis Pigmentosa), MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes), or MERRF syndromes (Myoclonic Epilepsy with Ragged-Red Fibers) (Bosbach et al., 2003; DiMauro and Schon, 2003; Kabunga et al., 2015).
In previously reported cases, several kinds of mtDNA deletions, ranging from 1.3 to 9.8 kb, were found in KSS patients (Klopstock et al., 1995; Pitceathly et al., 2011; Dominic et al., 2014; Kabunga et al., 2015; Maeda et al., 2016). Interestingly, the clinical symptoms and genetic mechanisms are distinct among these populations. For example, the most common 4.9 kb deletion located between positions 8470–13,447, which included several important genes, including MT-ATP8 (ATPase 8), MT-ATP6 (ATPase 6), MT-CO3 (COXIII), MT-ND3 (ND3), MT-ND4L (ND4L), MT-ND4 (ND4), MT-ND5 (ND5), and MT-ND6 (ND6), as well as tRNAGly, tRNAArg, tRNAHis, tRNASer(AGY), and tRNALeu(CUN) (Moraes et al., 1989). Seyrantepea et al. (1999) found that a 5-year-old girl with this deletion presented with failure to thrive, ptosis, ragged-red fibers, but not retinitis, abnormal renal, liver, or pancreatic functions. In another report, a novel mtDNA deletion (11,259–14,368) was identified in a 51-year-old KSS patient with slowly progressive ptosis, double vision, severe external ophthalmoplegia, and muscle weakness. There were no signs of cardiac dysfunction. The 3.1 kb-deletion covered MT-ND4 (ND4), MT-ND5 (ND5), MT-ND6 (ND6), as well as tRNAHis, tRNASer(AGY) and tRNALeu(CUN) (Klopstock et al., 1995). Thus, analysis mtDNA deletion in additional populations is still important to broaden the mutational spectrum of KSS disease and is helpful to understand the connection between mtDNA deletion and clinical symptoms.
In our study, we identified a novel 6600 bp mtDNA deletion in a pair of KSS twin boys. The novel mtDNA deletion located at the position between 8702 and 15,302 (Fig. 7). Through the genetic analysis, we found the deletion covered several genes encoding enzymes in the respiratory chain, such as MT-ATP6 (ATPase 6), MT-CO3 (COXIII), MT-ND3 (ND3), MT-ND4L (ND4L), MT-ND4 (ND4), MT-ND5 (ND5), MT-ND6 (ND6) and partially MT-CYB (Cyt b) as well as tRNAGly , tRNAArg , tRNAHis , tRNASer(AGY), tRNAGlu, and tRNALeu(CUN) . This kind of mtDNA deletions can affect specific proteins and disturb the function of the mitochondria and further cause pathological consequences. For example, the Cyt b gene encoded cytochrome b subunit of complex III (Fisher and Meunier, 2001). The partial deletion of Cyt b gene resulted in truncated cytochrome b and disrupted the function of the respiratory chain. COX III gene mutations would disturb the assembly of complex IV and further effect on the activity of cytochrome c oxidase (Schon et al., 1988). Since cytochrome c oxidase can be lethal in utero because there is no metabolic compensation, we predicted the 6600-bp deletion mtDNA and full-length mtDNA coexisted in the patients and the proportion of mutated to normal mtDNA determined the severity of clinical symptoms. It could explain why the clinical signs were more apparent in patient II-2 (with a degree of heteroplasmy of 93.7%) than patient II-1 (heteroplasmy level was 78.3%) in our study.
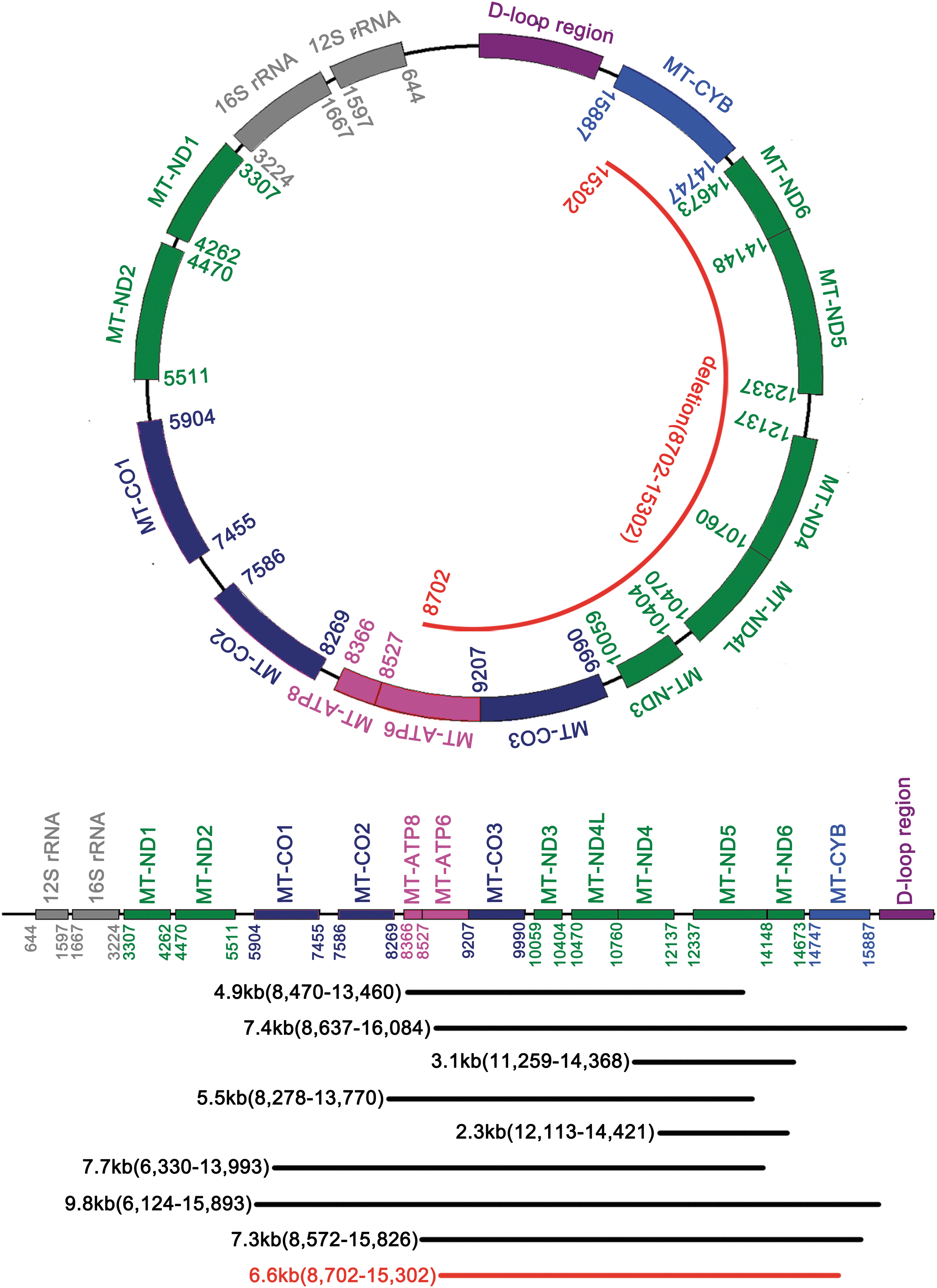
Localization of the novel deletion and known common deletion.
In conclusion, it is the first time to report the novel 6600 bps deletion on ChM: 8072–15,302 from a pair of twins affected with KSS. The rearrangement of large deletion would be the most likely reason for the disease. This finding may broaden the mutational spectrum of KSS disease and is helpful in understanding the correlation between genotype (mtDNA deletion) and phenotype (clinical symptoms) of KSS. However, the molecular mechanism underlying these complicated symptoms of KSS and whether the large deletion on mtDNA caused the clinical phenotype alone or in combinations with gene mutations on nuclear DNA still need to be studied. Further understanding of genotype/phenotype correlation of KSS might be beneficial for developing the treatment of KSS as well as other mitochondrial disorders.
Footnotes
Disclosure Statement
The authors declare that they have no competing interests.
Authors' Contributions
Luo Guo did the data curation. Haiting Ji carried out the experiments and analyzed the NGS data. Haiting Ji wrote the article. All authors read and approved the final version of the article.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (81600816).