
Abstract
Cervical squamous cell carcinoma (CESC) is a human papillomavirus-driven tumor that the underlying molecular mechanisms are unclear. This study aimed to elucidate the key candidate genes and potential mechanism in CESC by bioinformatics analysis. A total of 132 common differentially expressed genes (DEGs) were identified based on three expression profile data sets. A multivariate Cox proportional regression model was used to develop a four-gene prognostic signature. Mechanistically, the correlationship between MMP1 and tumor-infiltrating lymphocytes was further analyzed. Furthermore, annotations were investigated by Gene Ontology (GO) and The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses. Finally, potential drugs for CESC treatment were predicted by Connectivity Map. We profiled four genes (EFNA1, ANLN, MMP1, and ZWINT) with significant prognostic values for CESC. Multiple public available data sets were used for mRNA expression and prognostic characterization. Subsequently, GO and KEGG pathway analyses showed DEGs were mainly enriched in cell cycle, immunity, and metabolic-related pathway. We then conducted an integrated analysis of MMP1, and the expression of MMP1 showed significantly inverse association with the amount of CD8+ T cells, CD4+ T cells, and macrophages infiltration. Our findings suggest the four-gene signature may be associated with prognosis. We further revealed that MMP1 may be a novel biomarker for immunotherapy, and prognostic judgment of patients with cervical cancer.
Introduction
Cervical cancer remains one of the most common gynecologic malignancies, with >500,000 new cases and 300,000 deaths worldwide each year (Bray et al., 2018). The squamous cell carcinoma subtype is a main type, which accounts for 80–90% of cervical cancer cases (Printz, 2019). Ninety-five percent of the cases are caused by persistent infections with human papillomavirus (HPV), and HPV16 and HPV18 are the dominant subtypes (Schiffman et al., 2011). Although there were improvements in the diagnosis and therapy in the past decades, a significant number of patients were still diagnosed in late stage and could not get satisfactory clinical outcomes. Therefore, it is of great importance to further explore more effective prognostic biomarkers and therapeutic targets.
As HPV-driven cancer, cervical cancer appears at least partly mediated by the immune system (Komdeur et al., 2017). Checkpoint immunotherapy has shown significant effectiveness in lung, bladder, renal, and head and neck cancers (Otter et al., 2019). Cervical cancer will hopefully be within the expansion of the use of immunotherapeutic agents. However, the roles and mechanisms of how the interactions between tumor and the immune system impacting patients outcome remain unclear. Tumor-infiltrating lymphocytes (TILs) play an essential role in tumor-associated immune responses (Stanton et al., 2016). According to the specific antigens in the cell membrane, TILs could be divided into several subsets, and each of TIL subsets has different roles in the progression of the tumor. Many researchers have reported that TILs improved prognosis in various types of tumor, including colon, ovarian, lung, and breast cancer (Bremnes et al., 2016; Morris et al., 2018; Zhang et al., 2018; Truntzer et al., 2019). Of note, some studies have indicated a distinct correlation between different TIL intensities and different prognosis values (Wang et al., 2014; Vacchelli et al., 2015; Noble et al., 2016). The prognostic role of TILs is controversial currently; therefore, TILs have gained increasing attention in the treatment and prognosis prediction of tumors. Considering the considerable heterogeneity of TILs, the clinical impact of other immune cells in cervical squamous cell carcinoma (CESC) remains poorly understood.
This study aimed to identify useful signature genes to indicate poor clinical outcomes and investigate the correlations of tumor immunity with prognosis of CESC, which may assist us in evaluating therapeutic decisions and the prognosis for CESC patients. A signature of four genes was discovered and validated across the GEO cohorts, The Cancer Genome Atlas (TCGA) cohort, and public databases. The signature genes were not only predictive of clinical outcomes of CESC but also significantly associated with tumor immune. Furthermore, the most significant gene MMP1 was selected for further analysis. We investigated the correlation of MMP1 with tumor-infiltrating immune cells through Tumor Immune Estimation Resource (TIMER) and TISIDB. The results in this report revealed the critical role of MMP1 in CESC as well as provide an underlying mechanism and the potential relationship between MMP1 and tumor-immune interactions.
Materials and Methods
Gene Expression Omnibus data set and data processing
Gene expression raw microarray cell intensity (CEL) profiles of cervical cancer were evaluated in three independent data sets from the Gene Expression Omnibus database (accession nos. GSE63514, GSE6791, and GSE7410) (Pyeon et al., 2007; Biewenga et al., 2008; den Boon et al., 2015), which included 28 tumor tissue samples and 24 cancerous cervix samples; 20 tumor tissue samples and 8 cancerous cervix samples; 35 tumor tissue samples and 5 cancerous cervix samples, respectively. When more than one probe matched the same gene ID, the mean expression value of the gene was used for our study.
TCGA data set
The CESC data set containing 255 tumor samples and two normal samples, which included raw counts of RNAseq expression data and corresponding clinical information were obtained from TCGA data set. The TCGA data set was divided into three parts, two parts were selected randomly as a training cohort, and the complete data set was chosen as a validation cohort.
Identification of common differential expression genes
The GSE63514, GSE7410, and GSE6791 expression profiles were normalized and analyzed using the limma package (Smyth et al., 2005). The differential expression genes (DEGs) overlapping in the three cohorts were named as common DEGs. Furthermore, the featured genes were chosen based on the following criteria: (1) paired t-test analysis with p-value <0.05; (2) gene median expression in cervical cancer >0, and in adjacent normal tissue >0; (3) a two-tailed t-test was used to identify DEGs with the limma R package, and the |log2foldchange (log2FC)| >1 and adjusted p < 0.05 was set as the threshold to define the differences genes. All the data processing and normalization were performed using the R scripts.
Functional analysis of common DEGs
To identify their associated pathways and function annotations, Gene Ontology (GO) and The Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted using DAVID Bioinformatics Resources (Huang da et al., 2009). GO analyses classified the common DEGs into three categories, such as biological process (BP), cellular component (CC), and molecular function (MF). KEGG analyses were conducted to determine the pathways in which the DEGs were significantly enriched in p < 0.05 and enrichment score (ES) >1.0 were used as the cutoff significant criteria.
Identification and selection of prognosis-related genes
Cox's proportional hazards model is commonly employed in survival analysis. To assess the association between the expression of common DEGs and patients' overall survival, univariable Cox regression analysis was applied by the survival package and 10 significant related genes were selected out (p < 0.05). Therefore, as candidate variables, the 10 prognosis-related genes were further analyzed through stepwise multivariate Cox proportional hazards regression analysis. By fitting all candidate predictive models into Akaike information criterions (AICs), an optimal predictive model was selected as with the lowest AIC value, and the best performance efficacy predictive model was acquired. Subsequently, four genes (EFNA1, ANLN, MMP1, and ZWINT) were selected, and a four-gene-based prognostic model was established to evaluate the survival risk of each patient as follows:

Consequently, each of the four prognosis-related genes weighted by their estimated regression coefficients in the multivariable Cox regression analysis developed the risk score formula in both the training set and the validation set.
Risk score formula establishment and validation
To validate the four-gene risk signature in the internal validation data sets, we calculated the risk score for each patient in the complete TCGA cohort. The patients were then divided into a high-risk and a low-risk group based on the corresponding median risk score. The prediction accuracy of this risk model was determined by a time-dependent receiver operating characteristic (ROC) analysis. To compare the survival time difference between the low-risk and high-risk group, Kaplan–Meier curve was plotted by the survival package using the log-rank test.
Signature genes selected and validation
Furthermore, prognosis-related genes in CESC was validated using public database PROGgeneV2 (Goswami and Nakshatri, 2014), Gene Expression Profiling Interactive Analysis (GEPIA) database (Tang et al., 2017), and Oncomine database (Rhodes et al., 2004). PROGgeneV2 contains data from 134 cohorts from 21 cancer types, was used to test the survival differences in combined signature genes expression. The data deposited in GEPIA are retrieved from the UCSC Xena server, including 9736 tumor samples and 8587 normal samples, whereas ONCOMINE is a cancer microarray database that contains 65 gene expression data sets comprising nearly 48 million gene expression measurements from >4700 microarray experiments. These two databases were used to verify the prognostic signature of the four genes.
Gene set enrichment analysis identifies a high-risk group-related signaling pathway
Samples of the complete cohort were divided into a high-risk group and low-risk group depending on the expression of the four-signature genes. To identify the potential mechanism of four-gene prognosis signature, gene set enrichment analysis (GSEA) was performed by GSEA software version 3.0 (Subramanian et al., 2005). Annotated gene sets c2.cp.kegg.v5.2.symbols.gmt were chosen as the reference gene sets. false discovery rate (FDR) <0.25, and |ES| >0.8 were used to determine statistical significance, and set as the cutoff criteria.
Mining the immune-related mechanism of MMP1
The TISIDB database integrates 988 reported immune-related anti-tumor genes, high-throughput screening techniques, molecular profiling, and paracancerous multi-omics data, as well as various resources for immunological data retrieved from seven public databases (Ru et al., 2019). In this study, the TISIDB was employed to analyze the relationship between the clinical outcome and the expression levels of MMP1. Then, the correlations between MMP1 expression and TILs were investigated further. TIMER is a web resource for systematical evaluations of the clinical impact of different immune cells in diverse cancer types. The TIMER was used to explore the correlations between expression of MMP1 and each TILs (B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cell types) (Li et al., 2017). Purity-corrected partial Spearman method was selected. REACTOME is function analysis module in TISIDB, resource for understanding functions of genes from the molecular level, was used to exhibit the enrichment analysis pathways that involved of MMP1. p-Value <0.05 was set as the threshold value.
Related small molecule drugs screening
The common DEGs between CESC samples and adjacent normal samples were used to query the CMap database, which was used to predict potential small molecule drugs (Lamb et al., 2006). A positive connectivity score suggests that a drug can induce the input signature, whereas a negative connectivity score suggests that a drug can reverse the input signature in human cell lines, and the drugs with negative connectivity scores were identified as candidate molecules with potential therapeutic value. After sorting all instances, the connectivity score of various instances was filtered by the number of instances (N > 10) and p-value < 0.05, and seven small molecules were selected. The ES representing similarity were calculated (ranging from −1 to 1). As a result, a negative connectivity score drug was selected for it means that the drug can reverse the input signature in human cell lines and identified as candidate molecules with potential therapeutic value. Besides, tomographs of these candidate small molecular medicine were observed in the PubChem database.
Results
Identification of common DEGs and functional annotation
The overall flowchart of this study is summarized in Figure 1. After the analyses of GSE63514, GSE6791, and GSE7410 data sets, a total of 120 CESC samples (83 tumor tissues and 37 normal tissues) were explored. There were 3201 DEGs screened from the GSE63514 data set, including 2340 upregulated and 861 downregulated genes; 2811 DEGs screened from the GSE6791 data set, including 312 upregulated and 2499 downregulated genes; 843 DEGs selected from the GSE7410 data set, including 491 upregulated and 352 downregulated genes. The volcano plots of DEGs among each data set are shown in Figure 2A–C. Cluster analyses of DEGs showed two significantly different distribution patterns between the tumor tissue and normal samples, suggesting crucial roles of DEGs in tumorigenesis and progression of CESC (Fig. 2D–F). By Venn diagram analysis, 132 common DEGs in the intersection of the three data sets were identified and selected for further analysis, and the top 16 genes are shown in Figure 3B.
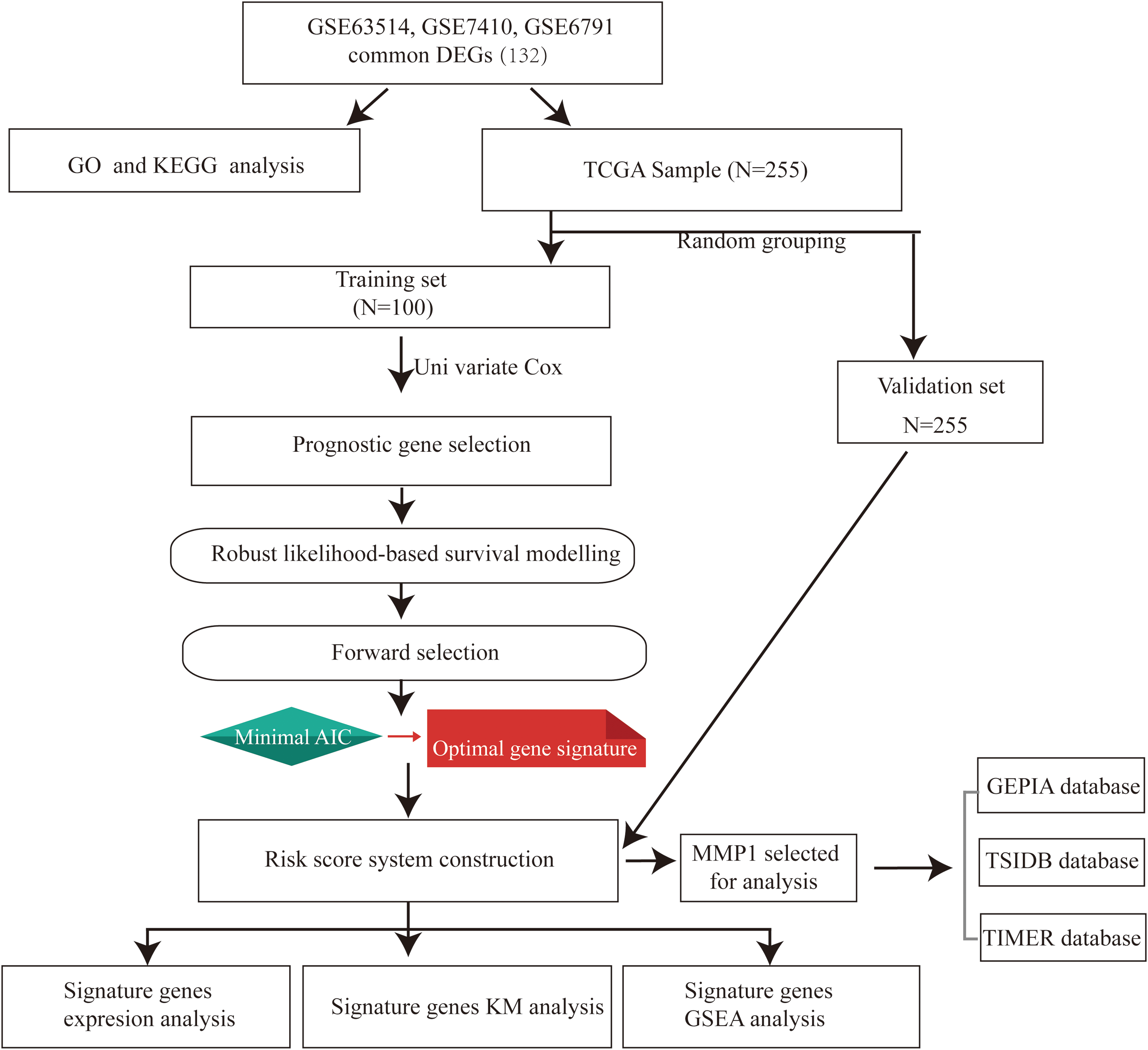
Flowchart for this study. AIC, Akaike information criterion; DEGs, differential expression genes; GEPIA, Gene Expression Profiling Interactive Analysis; GO, Gene Ontology; GSEA, gene set enrichment analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; KM, Kaplan–Meier; TCGA, The Cancer Genome Atlas; TIMER, Tumor Immune Estimation Resource. Color images are available online.
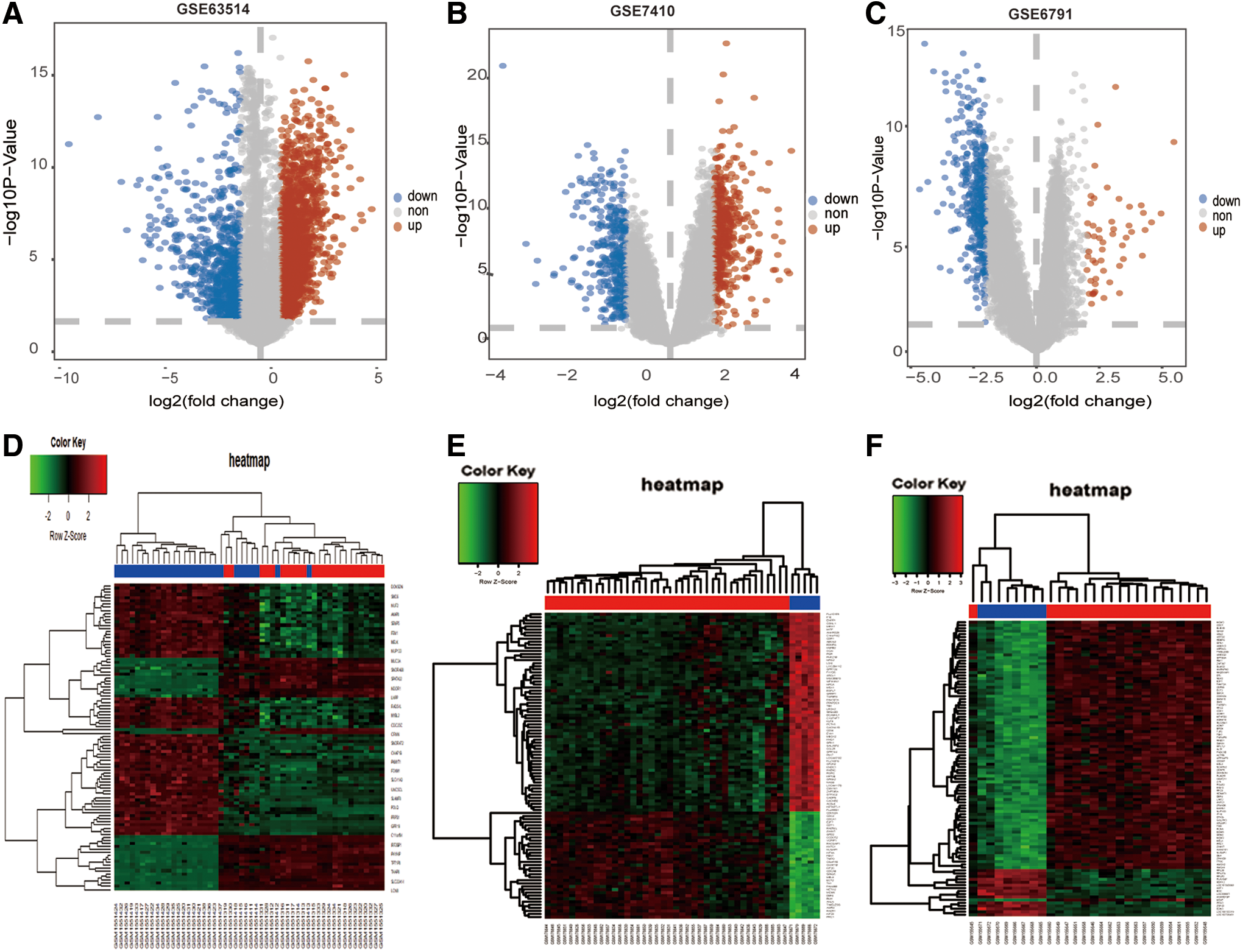
DEGs in three data sets.
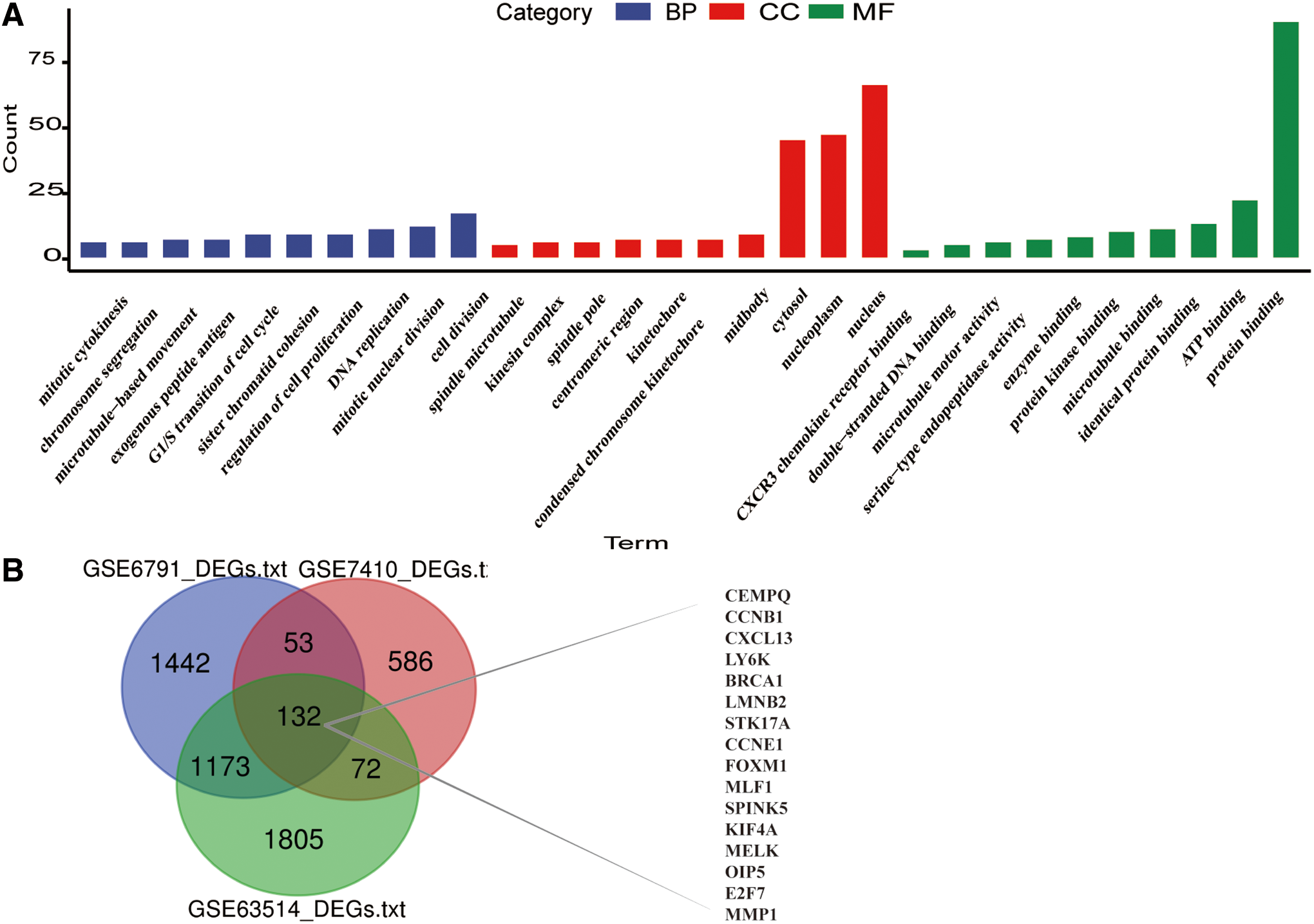
Functional annotation and KEGG pathway enrichment of common DEGs
GO analysis and KEGG pathway enrichment analysis were performed for the common DEGs to explore the potential BPs and function pathways associated with CESC. A total of 95 GO terms were significantly enriched (FDR <25%) (Supplementary Table S1). The top 10 GO terms in each functional category, including MF, BPs, and CC, are signified in Figure 3A. The GO terms majorly clustered in cell division, cell proliferation, cell cycle, immunity, and metabolic-related pathway, which are essential for rapid tumor growth (Supplementary Table S2). These results suggest that changes in immune and metabolic are necessary for tumorigenesis and development.
Moreover, the results of KEGG pathway analyses demonstrated that most of these target genes were enriched in cancer-related pathways, including p53 signaling pathway, transcriptional misregulation in cancer, and chemokine signaling pathway, indicating a role of the common DEGs in affecting tumorigenesis and progression of CESC (Table 1). We found that one gene may participate in multiple pathway, therefore, an intersection visualization plot was generated through UpSet plot (Fig. 4).

The Kyoto Encyclopedia of Genes and Genomes Pathway Enrichment Analysis of Common Differential Expression Genes
FDR, false discovery rate; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Signature genes showed the greatest prognostic value in patients with cervical tumors
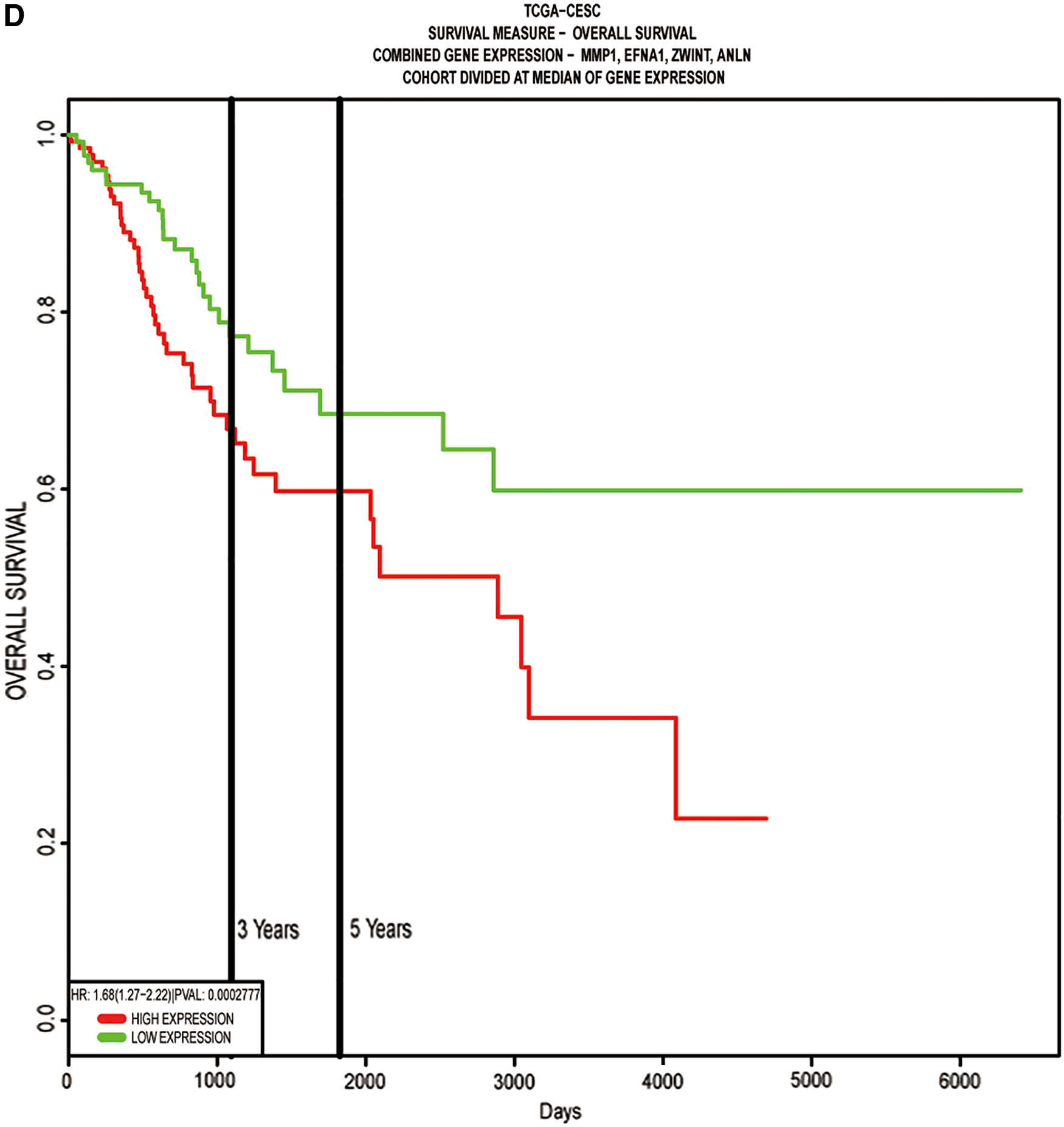
Given the significant association of common DEGs with tumorigenesis and development, we further investigate the potential roles in clinical outcomes. Univariate Cox proportional hazards regression revealed that EFNA1, ANLN, MMP1, TFRC, ZWINT, APOBEC3B, TRIM59, IFT80, FZD6, and PLOD2 were significant prognostic factors (Table 2). Multivariate Cox proportional hazards regression analysis of the 10 genes revealed a significant prognostic model comprising four different genes (EFNA1, ZWINT, ANLN, and MMP1). Among the four genes EFNA1, ANLN, and MMP1 were risk factors (hazard ratio (HR)>1), whereas ZWINT was identified as protective factor (HR <1). More information about these four genes is given in Table 3. The two ROCs exhibited high predictive abilities in both training cohort (n = 160) and validation cohort (n = 255) (Fig. 5A). The Kaplan–Meier analysis showed that patients in the high-risk group had a shorter overall survival time than patients in the low-risk group (p < 0.01; Fig. 5B, C).
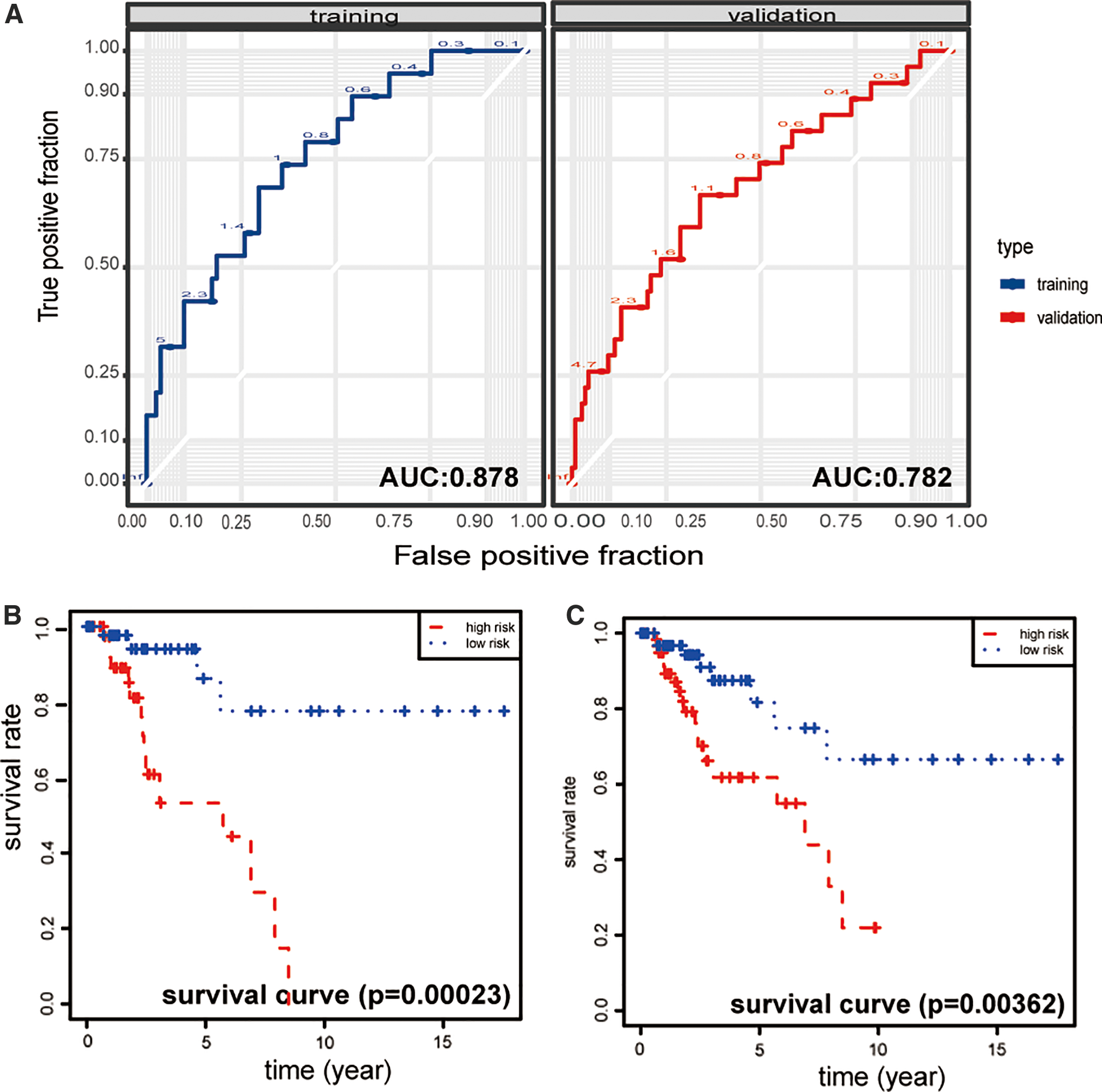
Validation the prognostic value of the four-gene signature.
The Detailed Information of the 10 Genes Significantly Associated with Overall Survival in Univariable Analyses
HR, hazard ratio.
The Four Prognosis-Associated Genes to Establish the Risk Score System
Bold font indicates p-value is less than 0.05.
CI, confidence interval.
Validation and analysis of signature genes
We further compared the survival difference between the high-risk group and the low-risk group in external database PROGgeneV2. The consistent results was obtained using from external verification that the high-risk group had had a shorter overall survival time (Fig. 5D, HR: 1.68 [1.27–2.22], p = 0.0002777). In addition, we analyzed the differential mRNA expression levels of EFNA1, ZWINT, ANLN, and MMP1 between cervical cancer and normal cervical tissues using the GEPIA database, which are generally consistent with those obtained from the Oncomine database (Fig. 6). Both databases showed that EFNA1, ZWINT, ANLN, and MMP1 mRNA expression levels were upregulated in cervical cancer compared with normal tissues, and indicated that EFNA1, ZWINT, ANLN, and MMP1 play important roles during the tumorigenesis and development of CESC.
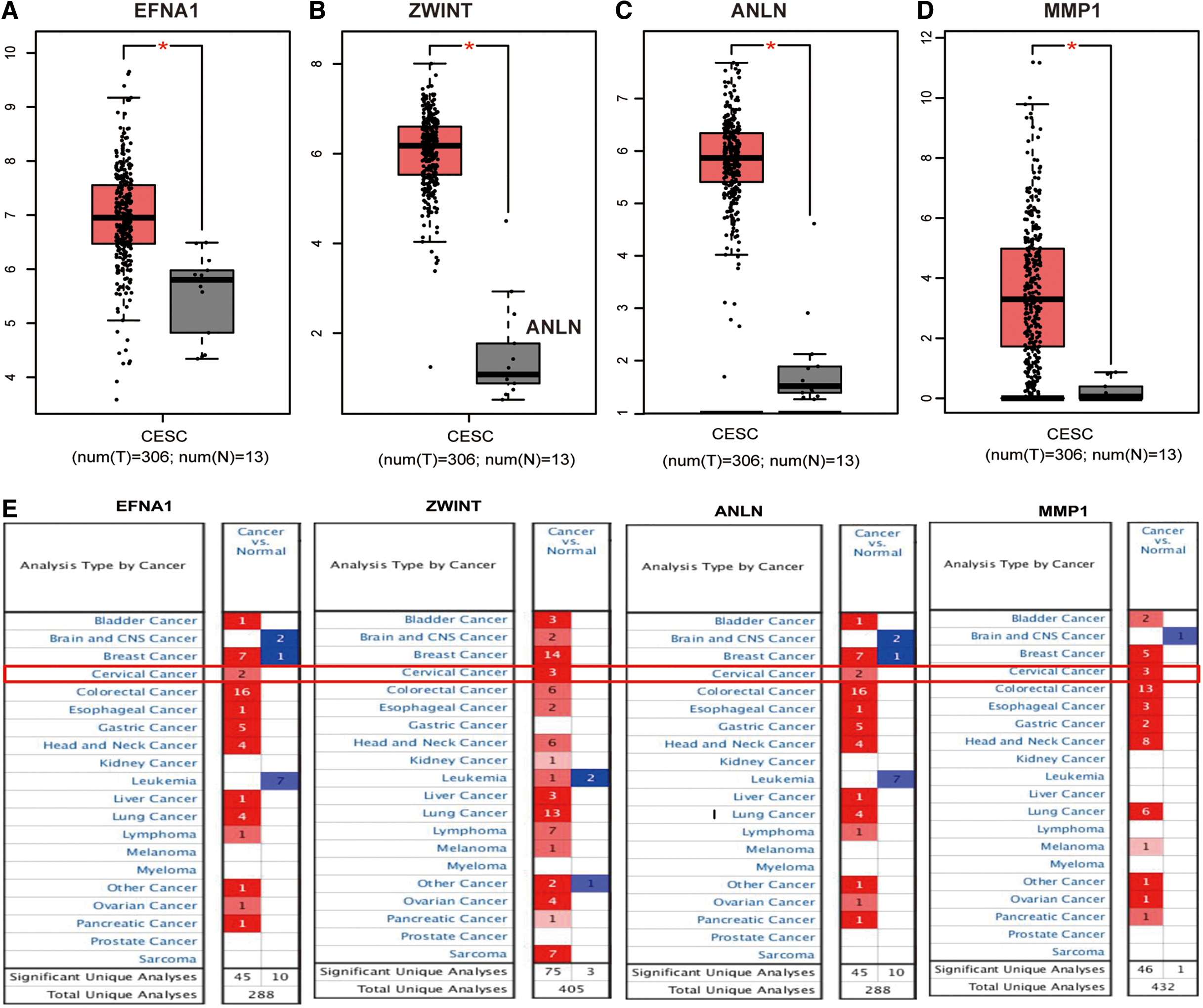
Validation of the gene expression levels of EFNA1, ZWINT, ANLN, and MMP1 between CESC tissues and normal cervical in GEPIA database.
Gene set enrichment analysis
Owing to such different prognostic outcomes between the high-risk and low-risk groups, we sought to investigate possible differences and molecular mechanisms by GSEA, which verified an enhanced antiapoptosis and epithelial-mesenchymal transition (EMT) signature in the high-risk group (Fig. 7). Therefore, we propose the existence of an intense regulatory role for the development and progression in high-risk cervical cancer patients.

GSEA results for high- vs. low-risk group
Regulation of immune molecules by MMP1
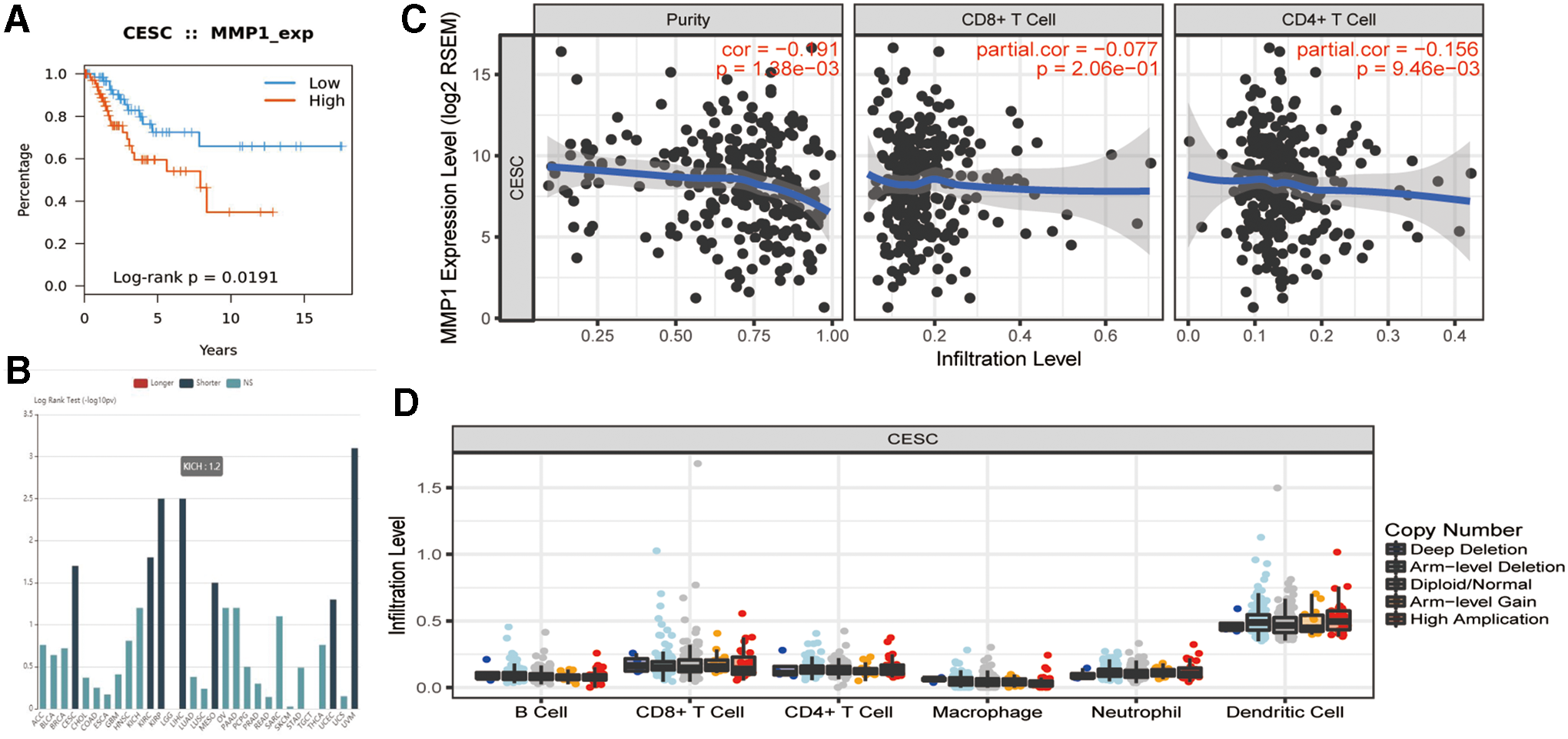
The KEGG pathway enrichment analysis showed that MMP1 is a hub gene of two cancer-related pathways. Besides, TISIDB-Reactome pathway analysis support suggested that MMP1 was involved in the cytokine signaling in the immune system (Table 4). Hence, we conducted an integrated analysis to predict the potential biological roles of MMP1 in tumor-immune of CESC. The results show that the expression of MMP1 was correlated with the abundance of TILs, whereas copy number does not appear to correlate with PD-L1 expression in cervical cancer (Fig. 8). More important, we found that patients with low expression of MMP1 exhibited significant treatment response of checkpoint immunotherapy (Table 5). To identify signatures and mechanisms of the relationship between MMP1 and response to checkpoint blockade (e.g., anti-PDL1 and anti-PD1), correlation analysis was performed, indicating that the expression of MMP1 is positively correlated with infiltration of macrophages, while negatively correlated with infiltration of CD8+ memory T and CD4+ memory T, which may be the reason why high MMP1 expression leads to better immune checkpoint treatment response (Figs. 8 and 9).

Reactome-MMP1
Points in the Scatter Plot Represent the Expression Difference of MMP1 in Various Data Sets (TISIDB)
See scatter plot, Fig. 8C.
CMap analysis
By CMap analyzed, we identified seven drugs with positive CMap ES of >0 and PCMap-value of <0.001 the most related small molecule drug trichostatin A with highly significant correlations and the maximum number of instances were identified. Trichostatin A, a histone deacetylase (HDAC) inhibitor, showed a significantly negative correlation and the tomography of trichostatin A was found in the PubChem database and shown in Figure 10. The screened small molecules are listed in Table 6.
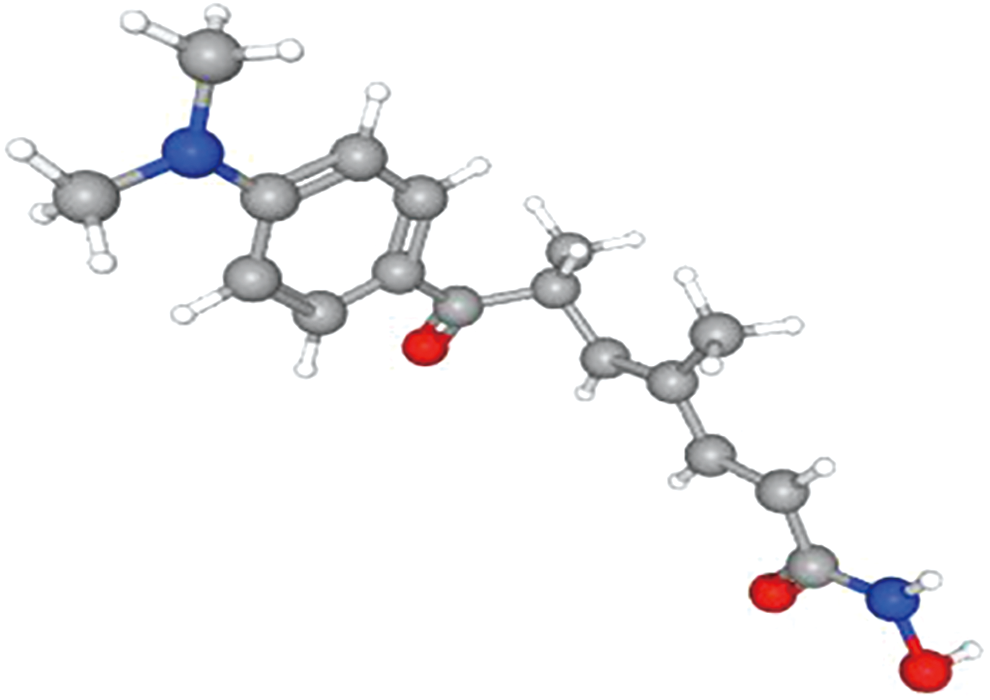
The tomographs of the candidate small molecule drug trichostatin A for CESC. Color images are available online.
The Top 10 CESC-Related Small Molecules with Highly Significant Correlations in Results of CMap Analysis
Drug in bold type is the most promising treatment small molecule drug identified in this study.
Discussion
In this study, we used comprehensive bioinformatics analyses to identify four prognosis-related signature genes (EFNA1, ZWINT, ANLN, and MMP1), the expression of which was significantly related to OS in CESC patients. In addition, the risk score of the four-gene signature demonstrated superior ability to divide CESC patients into high-risk and low-risk groups with significantly different OS in training data set and validation data set, as well as in Pan Cancer Prognostics Database (PROGgeneV2).
Furthermore, ROC curve analysis also suggested that these genes had an excellent value for predicting the OS for CESC patients. In the further GSEAs, we found that high-risk group showed an enhanced antiapoptosis and epithelial-mesenchymal pathway, implying that the four-gene signature may be involved in regulation of the prognosis of cervical cancer through antiapoptosis and the EMT pathways. Activation of EMT pathway promotes local tumor invasion and tumor cell metastasis in various cancers (De Craene and Berx, 2013; Jung et al., 2015). Qureshi et al. (2015) have reported that the EMT plays a vital role in cervical cancer progression and metastasis. In addition, previous studies have demonstrated that activation of EMT pathway promotes lymph node (LN) metastasis in cervical cancer (Noordhuis et al., 2011). Taken together, these results suggest that the high-risk populations may have a worse prognosis because EMT pathway activation promotes cervical cancer progression, which contributed to the poor prognosis of CESC.
The signature gene MMP1 was identified as biomarkers in lung adenocarcinoma, esophageal cancer, bladder cancer (Wieczorek et al., 2015; Liu et al., 2016; Ni and Sun 2019). ANLN was reported as a prognostic marker for head and neck squamous cell carcinoma (Shimizu et al., 2007). However, no previous study has considered the possible role of EFNA1, ZWINT in cancer. We first proposed two new prognostic markers for cervical cancer. Therefore, the signature may be a potential good predictor of prognosis for cervical cancer patients. This research provides more deep insights into possible molecular mechanisms of prognosis for CESC. However, all of these four genes need further experimental verification to demonstrate their roles in CESC.
Significantly, MMP1 was chose for subsequent in-depth investigations for its correlations with tumor immunity and possible mechanism. Finally, we selected a small molecule drug that has the potential values to treat CESC by CMap database.
The mRNA expression level of the four genes was significantly upregulated in cervical cancer compared with normal, both revealed in silico and Oncomine database analyses. Patients with high EFNA1, ZWINT, ANLN, and MMP1 expression had significantly shorter survival than patients with lower expression of these genes. Moreover, the signature was confirmed to harbor high predictive value of CESC both in the validation cohort and the online databases PROGgeneV2. EFNA1 has been found as key serum markers for the detection of hepatocellular carcinoma development and progression, but no reporter in literature in CESC to date (Tian et al., 2010). Previously, Suman and Mishra (2018) reported that ZWINT were independent prognostic factors. Mechanistically, ANLN may be responsible for progressing cervical neoplasia to cervical cancer at an early stage (Xia et al., 2018). MMP1 was considered having a pivotal role in the regulation of cervical tumor growth and LN metastasis through EMT (Tian et al., 2019). These results are consistent with ours, and suggest that the four signature genes had an excellent value for predicting the 5-year survival prognosis for CESC.
To further explore the potential mechanism of the four genes, we observe that high-risk phenotype was associated with apoptosis pathway and EMT by GSEA. Several researchers had reported that EMT is the most important mechanism that plays a crucial role in the metastasis and invasion of cervical cancer (Feng et al., 2019; Li et al., 2019; Zeng et al., 2019) These results represent candidate biomarkers for predicting prognosis of CESC patients.
Given the prognostic role of MMP1 expression in CESC has been reported in several studies, its role in cervical cancer the immune modulation remains unstudied (Zhang and Zhao, 2016; Roszik et al., 2018; Tian et al., 2018). The KEGG pathway enrichment analysis showed that MMP1 is a hub gene of two cancer-related pathways. Besides, TISIDB-Reactome pathway analysis support suggested that MMP1 was involved in the cytokine signaling in immune system. Hence, we conducted an integrated analysis to predict the potential biological roles of MMP1 in tumor-immune of CESC.
In further study, we found out that there was a significant difference in the expression level of MMP1 between responders and nonresponders of PD-1 in four clinical immunotherapy researches. Indeed, low expression of MMP1 with better immunotherapy response rate. In addition, correlation analysis reported that the expression of MMP1 was correlated with the abundance of TILs, whereas copy number does not appear to correlate with PD-L1 expression in cervical cancer. PD-L1 immunohistochemistry alone is not yet an adequate biomarker for routine clinical use in deciding which patients to offer anti-PD-1 or anti-PD-L1 therapy. It has recently confirmed that baseline tumor-infiltrating lymphocyte status could also serve as a predictive biomarker for checkpoint inhibitor immunotherapy (Otter et al., 2016). Otter et al. (2019) suggested that the best predictive marker of response to PD-1 blocking agents in pretreatment biopsies was CD8+ memory T cell density at the invasive margin. In fact, the CD8+ memory T cell subset, displayed an activated and proliferative state and shown to be crucial for effective in vivo PD-1 blockade (Heeren et al., 2019). Therefore, CD8+ T cell density was more predictive of the response to treatment of PD-1 and PD-L1 blocking agents. However, no report has demonstrated a correlation between MMP1 expression and CD8+ memory in CESC. In this study, it is suggested that low expressed MMP1 may be crucial for evoking the enrichment of the effective (active) CD8+ memory T. In this study, we have for the first time revealed this inverse correlation between MMP1 and CD8+ memory T cells.
In contrast, CD4+ memory T cells have been shown to predict prognosis better than TNM (tumor-node-metastasis) classification system in colorectal cancer (Mlecnik et al., 2011). Moreover, recent reports have suggested that also showed a significant correlation between circulating highly differentiated memory CD4 T cells and objective clinical responses. Patients with high percentages of these memory subsets within the CD4 cell population also had about 50% response rates (Zuazo et al., 2018, 2019). Interestingly, patients with high memory CD4 T cells that also had lower percentages of PD-L1+ CD11b+ cells did not show objective responses. These results suggest that combining key systemic cellular parameters could be used to stratify patients with a high probability of responding to PD-L1/PD-1 blockade. It is likely that PD-L1 blockade in these cells may enhance their antigen presentation capacities, and together with memory CD4 T cells trigger strong antitumor activities. Consistently, PD-L1 expression in systemic immune cell populations was identified as a potential predictive biomarker of responses to PD-L1/PD-1 blockade therapy in lung cancer (Bocanegra et al., 2019). These results suggest that a combined memory CD4 T cells and expression of PD-L1 may be beneficial biomarkers. In fact, our results complemented the immune environment regulation values of MMP1 in CESC with multidimensional analysis.
Macrophages are one of the most abundant immune cells in the tumor microenvironment of solid tumors, and their presence correlates with reduced survival in most cancers (Nielsen and Schmid, 2017). Tumor-associated macrophages (TAMs) display several protumorigenic functions that have essential roles in cancer development and progression such as the ability to provide cytokines and induce tumor angiogenesis (Grivennikov et al., 2010). TAMs are a source of tumor-promoting interleukin (IL)-6 in several murine tumor models. Promotes colon tumor cell proliferation and protection from apoptosis through activation of signal transducers and activator of 66 transcription-3 (STAT3) (Bollrath et al., 2009; Grivennikov et al., 2009; Shrivastava et al., 2013). The poor prognosis of CESC caused by high expression of MMP1 may be related to the massive infiltration of TAM. Furthermore, through such a STAT3 activation process, CESC patients with high MMP1 expression may lead to advanced disease and present a higher risk for poor outcome, which becomes the basis for developing prognosis prediction biomarkers.
Furthermore, GSVA (TISIDB-Reactome) analysis demonstrated that the BPs of the genes were mainly associated with immune suppression, such as infection by HPV, cytokine signaling in immune system, IL-4 and IL-13 signaling. Previous studies indicated that STAT3, one of STAT family members, has been reported to be activated by inflammatory cytokines such as IL-4 and IL-13 in 68 integral signaling pathways (Lesina et al., 2011). IL-4 and IL-13 were involved in the STAT3 pathway, indicating that MMP1 may control macrophages to activate the STAT3 pathway to promote tumor progression. Collectively, these results demonstrated that MMP1 is a high candidate as a biomarker for immunotherapy and prognostic judgments of cervical cancer.
Revealing the relationship between MMP1 and immune infiltrating cells and identifying MMP1 as an essential prognostic factor are the highlights of this study. We are currently expanding these analyses during immunotherapy treatments to evaluate the dynamic changes of these cervical cancer populations in responders and progressors. Considering both the immune and gene expression profiling data provided by our study, we feel MMP1 is not only a potential prognostic biomarker but also a high priority consideration for predicting the efficacy of PD-1/PD-L1 targeted therapy. Furthermore, to identify more potential drugs for CESC treatment based on the common DEG profiles, CMap analysis was conducted and 18 candidate drugs (p < 0.001) were obtained from the CMap data set.
Moreover, we identified several small molecules with potential therapeutic efficacy against CESC. Trichostatin A (connectivity score of −0.788) was particularly interesting and it is considered to be the most important small molecule in our screening for the most research data supporting. Trichostatin A is a HDAC inhibitor (HDACi). HDACi, a novel class of anticancer drug, has been recently reported to suppress host immunity. Many studies demonstrated that trichostatin A may be a useful therapeutic tool for the reversion of EMT in various types of cancers, including colon cancer (Huang et al., 2019) and breast cancer (Vigushin et al., 2001). Reasonably it is presumed that the potential anticancer effect of trichostatin A could be further characterized and validated in further study.
Limitation
We would like to acknowledge some of the limitations of this study. First, this is a retrospective study; all the data of this study were obtained from publicly available databases. Second, further studies, including in vivo and in vitro experiments, are required to confirm the results. Third, the exact mechanisms of action of the four genes in CESC remain to be fully elucidated, and the molecular mechanism of MMP1 regulating the TILs infiltration will be another research point for us. Despite this limitation, these findings present an opportunity to rationally approach future combination immunotherapy trials in the treatment of recurrent cervical cancer. These discoveries suggest that a comprehensive investigation of tumor and immune cell interplay would assist both the elucidation of cervical cancer pathogenesis and the development of novel immunotherapy strategies.
Conclusion
In this study we identified a four-gene signature that may prove to be associated with progression and prognosis, which can provide the favorable prognostic value based on these four gene expression levels, and a candidate small molecule drug was identified, which provides direction for CESC targeted therapy. With further exploration, our data revealed that MMP1 was significantly highly expressed in CESC tissues and that high MMP1 expression was significantly correlated with poor prognosis TILs. Therefore, MMP1 may prove to be a novel biomarker for immunotherapy and prognostic judgment of patients with cervical cancer.
Footnotes
Acknowledgments
We would like to acknowledge the KEGG database developed by Kanehisa Laboratories. We also would like to acknowledge the Oncomine, GEPIA, TISIDB, TIMER, and TCGA databases for free use.
Authors' Contributions
M.Y. designed the study, as well as designed the figures and tables. S.Z. contributed to the statistical analysis, as well as wrote and corrected the article. All authors read and approved the final article.
Ethics Approval and Consent to Participate
Not applicable.
Patient Consent for Publication
Not applicable.
Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This study was supported by Natural Science Foundation of Liaoning Province (Grant No. 20180550781).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.