
Abstract
Prostate cancer (PCa) is a common malignant tumor in elderly men worldwide. Most primary PCas inevitably progress into castration-resistant prostate cancer (CRPC) after androgen deprivation therapy. The mechanisms contributing to this progression are still controversial. In this study, functional module genes, DNA methylations, core regulators, and potential drugs in primary PCa and CRPC were explored by integrating a series of bioinformatics analyses. First, 588 differentially expressed genes (DEGs) were identified. Combined with related genes, protein-protein interaction networks were constructed, and 22 and 14 significant modules were identified in primary PCa and CRPC, respectively. More DEGs were identified in differentially methylated genes in CRPC modules. The hub genes in CRPC included CDC20 and CDK1. Moreover, core noncoding RNAs and transcription factors that significantly regulate CRPC modules were identified, including TUG1, MALAT1, E2F3, and MED1. Finally, the prediction of potential drugs for primary PCa and CRPC was also performed. Exisulind and phosphodiesterase-4 inhibitors were predicted as potential drugs for CRPC. The results of this study provide a new way for biologists and pharmacists to understand the potential molecular mechanisms of CRPC and also provide valuable references for drug redirection and new drug development for PCa.
Introduction
Prostate cancer (PCa) is the most frequently diagnosed cancer in men, and although it has decreased in number in recent years, it still accounts for nearly one in five new diagnoses. It is the second leading cause of cancer-related mortality in males, accounting for ∼10% of all cancer deaths (Siegel et al., 2019). Androgen deprivation therapy (ADT) is the key treatment for locally advanced or metastatic PCa. However, in most patients, PCa regresses upon ADT, but inevitably relapses and becomes resistant to ADT, leading to castration-resistant prostate cancer (CRPC) (Debes and Tindall, 2004).
Multiple mechanisms have been proposed to explain the progression of PCa from hormone dependence to castration resistance (Pienta and Bradley, 2006). Although previous studies have shown that the abnormal expression of some specific genes contributes to CRPC progression, we should note that PCa, as a complex disease, especially in its castration-resistant stage, is rarely a consequence of the abnormality of a single gene and is usually the result of complex interactions and regulations involving large sets of genes. These genes interact with each other and are involved in a protein-protein interaction (PPI) network. Based on the PPI network, significant functional modules in PCa could be identified. Further bioinformatics analysis would help to identify the core genes and potential drugs for PCa, especially for CRPC. However, few such studies based on bioinformatics analysis in PCa were reported.
In this study, we performed a comprehensive analysis on the expression profiles of primary PCa and CRPC to screen differentially expressed genes (DEGs). Then, combined with known PCa-related genes, significant functional modules, pathways, and hub genes were identified and compared from the PPI network. Related DNA methylation, key transcription factors (TFs), and noncoding RNAs (ncRNAs) that may regulate significant functional modules were also analyzed. Moreover, potential drugs for primary PCa and CRPC were predicted based on drug-module interactions (Fig. 1). This study could provide useful insights for understanding the molecular mechanisms of CRPC and exploring potential therapeutic drugs at the gene level.

Flow diagram showing an overview of the study.
Materials and Methods
Data source and preprocessing
To identify DEGs between primary PCa and CRPC tissue, the gene expression profiles were searched in Gene Expression Omnibus (GEO); database. GSE32269 dataset with both two categories of tissues and balanced sample size was selected as data source for further analysis. GSE32269 was based on a GPL96 (HG-U133A) Affymetrix Human Genome U133A Array platform, and the expression files were deposited by Steven P. Balk's Lab (Cai et al., 2013). The microarray data of mRNA profiles included 22 hormone-dependent primary PCa (GSM799468-GSM799489) and 29 metastatic CRPC (GSM799490-GSM799518) samples.
The DNA methylation profiling data in primary PCa and CRPC were also downloaded from GEO (GSE41701 dataset). GSE41701 was high-throughput profiling of the DNA methylome by Enhanced Reduced Representation Bisulfite Sequencing, containing seven primary PCa (GSM1022921-GSM1022927) and six CRPC (GSM1022928-GSM1022933) samples (Lin et al., 2013).
To ensure comprehensive analysis, genes related to primary PCa (keywords: primary prostate cancer NOT castration-resistant prostate cancer) and CRPC (keywords: castration-resistant PCa) were searched in the National Center for Biotechnology Information (NCBI)-gene database and the Online Mendelian Inheritance in Man (OMIM); database.
Identification of DEGs and differentially methylated genes
The raw microarray expression data of mRNAs downloaded as Series Matrix files from the GEO database were mapped to the corresponding genes according to the SOFT formatted family files from the GEO database. The linear models for microarray data (Limma) package in R software were used to analyze the differentially expressed mRNAs between primary PCa and CRPC (Ritchie et al., 2015). All genes with |log fold change (logFC)| >1 and p < 0.05 were selected as DEGs for further study.
For the DNA methylation data, methylKit package in R software was used to identify the differentially methylated position (DMP) between primary Pca and CRPC (Akalin et al., 2012). Then, according to their locations in the genome region, all DMPs were annotated to the corresponding genes, which were defined as differentially methylated genes (DMGs).
PPI network construction and module selection
PPI networks are increasingly being used for identifying the interactions between proteins in various organisms. The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING), an online database resource provides uniquely comprehensive information for assembling, evaluating, and disseminating PPI networks in a user-friendly manner (von Mering et al., 2003). Cytoscape is a biological graph visualization tool for integrated models of biomolecular interaction networks (Smoot et al., 2011). Clustering with overlap neighborhood expansion (ClusterONE) is a complex recognition algorithm for mining overlapping densely connected regions from weighted or nonweighted PPI networks. It can be used to identify important local structures in various biological networks (Nepusz et al., 2012). In this study, STRING was used to analyze the PPIs of primary PCa and CRPC, which were then visualized with Cytoscape software. Moreover, based on the connectivity degree, the functional modules of the PPI networks in primary PCa and CRPC were analyzed by the Cytoscape plug-in ClusterONE.
Functional enrichment analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) is an online tool for systematically extracting biological meaning and functional annotations behind large gene or protein lists (Huang da et al., 2009). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis were performed by DAVID for the functional enrichment analysis of the modules in primary PCa and CRPC, especially for CRPC-specific modules. A p-value of <0.01 was chosen as the significance threshold.
Pivot analysis
The pivot node refers to (1) at least two interaction pairs with module genes and (2) the p-value in the significance analysis of the interaction between the node and each module being ≤0.05, with the statistical method being a hypergeometric test.
The pivot analysis for ncRNAs was as follows: the ncRNA-mRNA interactions in the RAID 2.0 (Yi et al., 2017) database were used as the background of interaction, and the interaction pairs between all ncRNAs and module genes were counted. Then, the interaction pairs between each ncRNA and module gene or other genes were counted. The ncRNA-module interactions were screened based on the p-value in the hypergeometry test (p < 0.05).
The pivot analysis for TF regulation was as follows: the human TF-mRNA regulatory relationships in the TRRUST v2 (Han et al., 2018) database were used as the background of interaction, and the interaction pairs between all TFs and module genes were counted. Then, the interaction pairs between each TF and gene in the module or other genes were counted. The TF-module interactions were screened based on the p-value in the hypergeometry test (p < 0.05).
The pivot analysis for potential drugs was as follows: the human drug-mRNA regulatory relationships in the DrugBank 4.0 database were used as the interaction background (Law et al., 2014). All drug-module interaction pairs were counted. Then, the interaction pairs between each drug and module gene or other genes were counted. Potential drugs were determined based on the p-value in the hypergeometry test (p < 0.05).
Verification of key genes by reverse transcriptase-quantitative polymerase chain reaction
PCa tissue samples were collected from ten patients with primary PCa and eight patients with CRPC by transrectal ultrasound-guided prostate biopsy, confirmed by experienced pathologists. All human samples were collected according to the Declaration of Helsinki, and informed consent was obtained from all patients. This study was approved by the Ethics Committee of the First Hospital of Jilin University and carried out in accordance with their regulations (Project identification code 2019-242). The mRNA expression level of the hub genes was detected by reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR).
Total RNA was extracted from fresh tissue with TRIzol (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA) and transcribed into complementary DNA (cDNA) using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, Inc.). Two-step qPCR was performed with SYBR Green PCR Master Mix on an ABI Prism 7700 SDS. The qPCR program began with the initial 3-min denaturation step at 95°C to activate the hot-start iTaqTM DNA polymerase, followed by 45 cycles of denaturation at 95°C for 10 s and annealing and extension at 60°C for 45 s. Results were normalized to GAPDH level. The following primers were used: polo-like kinase 1 (PLK1) (forward: 5′-CGAGGACAACGACTTCGTGT-3′, reverse: 5′-GGTTGCCAGTCCAAAATCCC-3′), CDC20 (forward: 5′-AGATGGACGACATTTGGCCA-3′, reverse: 5′-ATTGGACTGCCAGGGACACC-3′), CDK1 (forward: 5′-TGGATCTGAAGAAATACTTGGATTCTA-3′, reverse: 5′-CAATCCCCTGTAGGATTTGG-3′), and BUB1B (forward: 5′-CTTAGGGTGCAGCTGGATGT-3′, reverse: 5′-ACCCATCCCAGAAGACCTGT-3′). β-actin as an internal control (forward: 5′-GGGAAATCGTGCGTGACATTAAGG-3′, reverse: 5′-TAAAATAAATACACAACCCTCCT-3′).
Results
Identification of DEGs
Based on the Limma package in R language, 588 DEGs were identified between 22 primary PCa samples and 29 CRPC samples from GSE32269 (|logFC| > 1, p < 0.05) (Supplementary Table S1). Among these genes, 353 genes were upregulated and 235 genes were downregulated. The expression profiles of the DEGs in the two groups are shown in Figure 2.
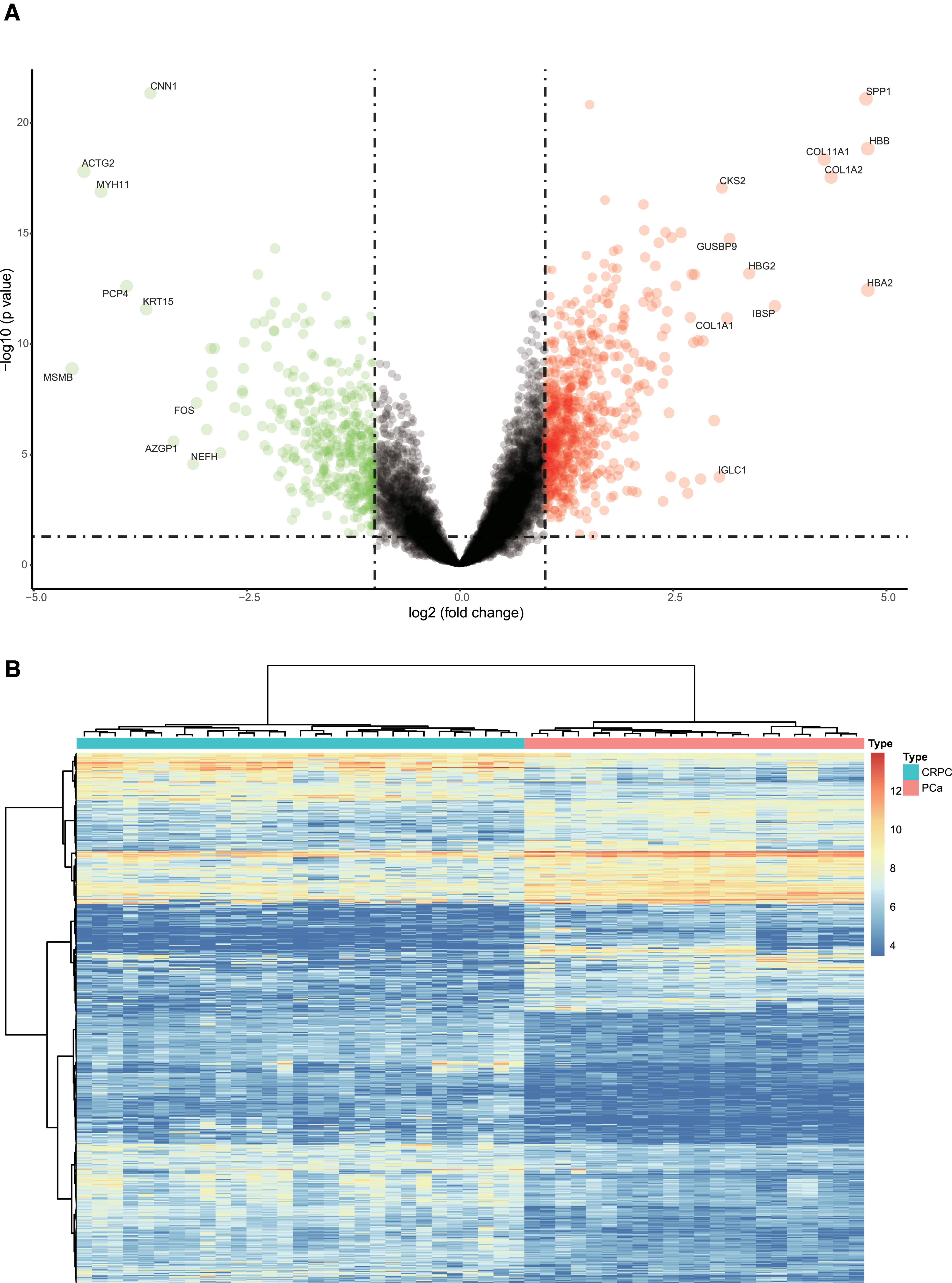
The volcano plot and the hierarchical clustering heatmap of DEGs between primary PCa and CRPC.
PPI network construction
To construct the PPI network of primary PC and CRPC, in addition to DEGs, related genes were also searched in the NCBI-gene database and OMIM database. There were two lists of 2457 genes and 200 genes related to primary PCa downloaded from the NCBI-gene database and OMIM database, respectively. For CRPC, there were two lists of 197 genes and 6 genes downloaded from the NCBI-gene database and OMIM database, respectively. Then, these lists of genes were combined with the identified 588 DEGs to construct the PPI network (Fig. 3). The combined number of genes for constructing the PPI network in primary PCa was 2957, while the combined number in CRPC was 758 (Supplementary Table S2). According to the STRING data, PPI networks of the primary PCa and CRPC genes were generated (Supplementary Fig. S1); a total of 205 and 51 clusters were identified in the two groups, respectively (Supplementary Table S3).

Venn diagrams showing the number of genes expressed as common, unique, and combined among the identified DEGs, genes in the NCBI-gene database, and genes in the OMIM database for primary PCa
Functional module analysis
Based on the PPI network, 22 and 14 significant modules were identified from the primary PCa and CRPC networks, respectively, by ClusterONE (minimum density ≥0.5, degree ≥3, and p < = 0.05) (Fig. 4). To determine active hub genes in the PPI subnetwork in primary PCa and CRPC, the connectivity degrees of the genes in the two groups of modules were calculated and ranked (Supplementary Table S4). The top-ranked genes were thought to be active hub genes in primary PCa and CRPC. The top six active hub genes in the primary PCa modules (connectivity degree ≥75) were lysophosphatidic acid receptor 1 (LPAR1), LPAR2, LPAR3, serum amyloid A1 (SAA1), Annexin A1 (ANXA1), and calcium-sensing receptor (CASR). The top five active hub genes in the CRPC subnetwork (connectivity degree ≥16) were PLK1, CDC20, CDK1, BUB1B, and MAD2L1. Expressions of some hub genes in CRPC were verified by RT-qPCR in PCa tissues (Fig. 5).
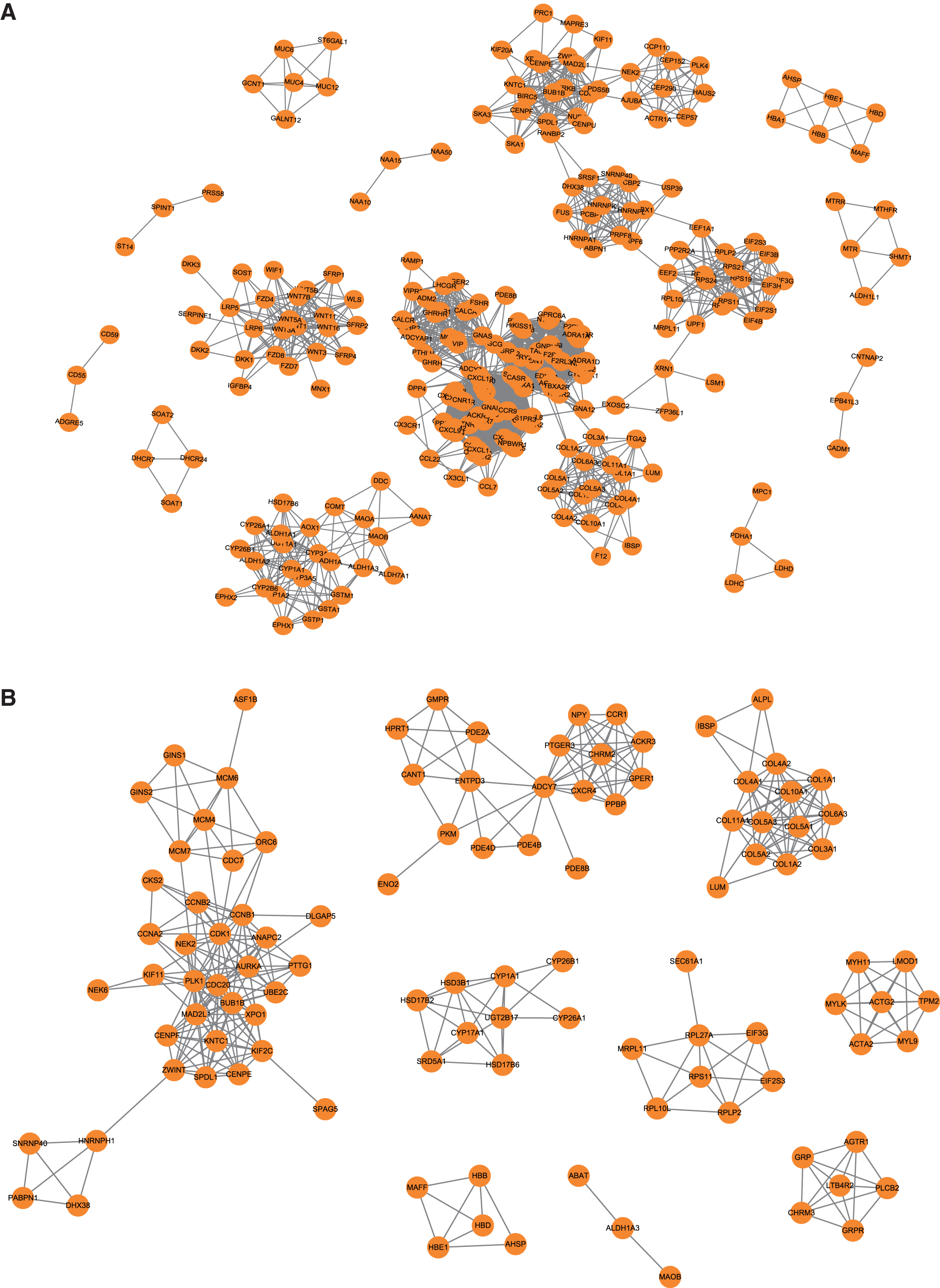
Twenty-two and 14 significant modules in the protein-protein interaction network of primary PCa
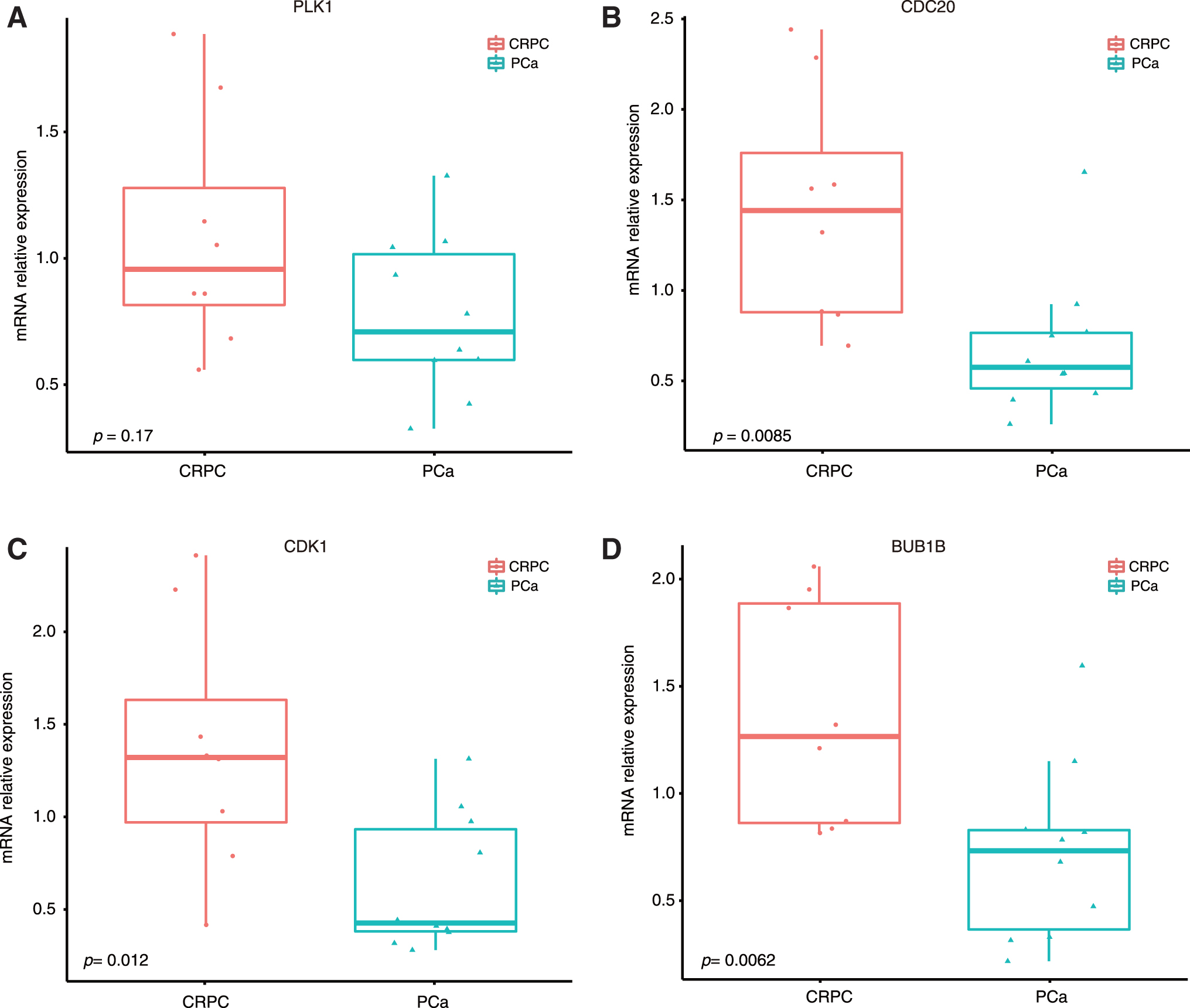
mRNA expression of top identified CRPC-related hub genes in clinical samples.
Functional and pathway analyses
Exploring the functions and pathways involved in the significant modules could help to elucidate the roles of dysregulated genes and their mechanisms. To determine and compare the biological functions of the module genes in primary PCa and CRPC, GO functional and KEGG pathway enrichment analyses were performed. The analyses identified 903 biological process (BP) terms, 55 cellular component (CC) terms, 91 molecular function (MF) terms, and 39 KEGG pathways in the 22 primary PCa modules, while 305 BP terms, 56 CC terms, 48 MF terms, and 20 KEGG pathways were identified in the 14 CRPC modules (Fig. 6 and Supplementary Table S5). The analysis showed that the function of genes in the primary PCa and CRPC modules was distinct.
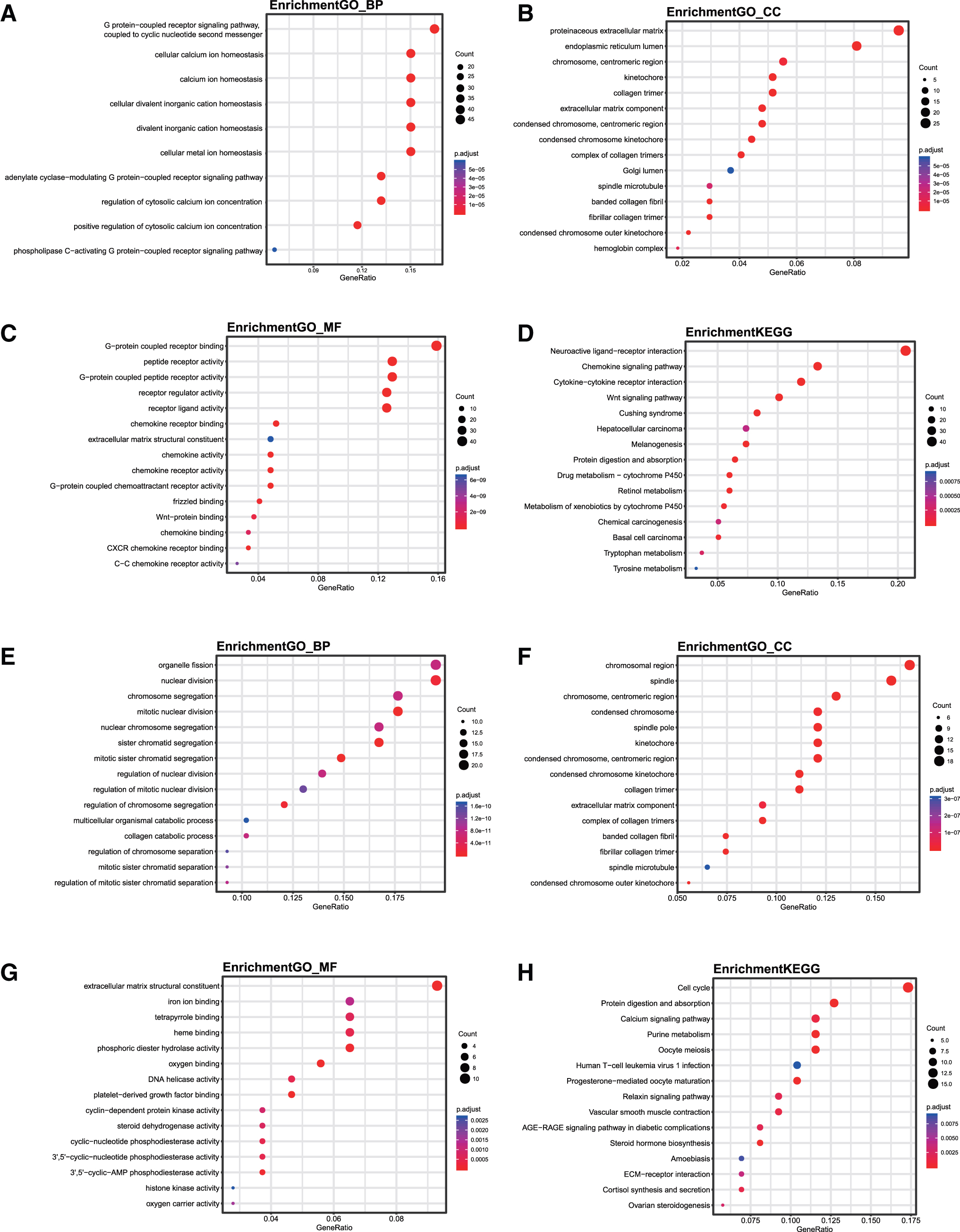
The functions and pathways of module genes identified in primary PCa and CRPC.
KEGG pathway analysis suggested that compared with that in the primary PCa modules, genes in the CRPC modules were involved in BPs, including the cell cycle, the calcium signaling pathway, and steroid hormone biosynthesis. Moreover, to clarify the functional changes in genes during CRPC progression, the specific module genes in CRPC were obtained by comparing the primary PCa and CRPC modules, and then GO functions and KEGG pathway analyses were performed for these specific module genes. A total of 308 BP terms, 48 CC terms, 33 MF terms, and 9 KEGG pathways were identified (Supplementary Table S6 and Supplementary Fig. S2). KEGG pathway analysis suggested that the DNA replication pathway was also enriched in CRPC-specific module genes.
Identification of DMGs in functional modules
From GSE41701 dataset, 149966 DMPs were identified between primary PCa and CRPC by the methylKit R package (|methylated difference| >25%, qvalue <0.05). These DMPs were annotated to 10,109 genes, including protein-coding genes, lncRNAs, and miRNAs (Supplementary Table S7). Then we analyzed the DNA methylated states in significant modules in primary PCa and CRPC. In primary PCa module, there were 133 DMGs identified, while 48 DMGs were identified in CRPC module (Supplementary Table S8). The percentages of DMGs in both modules were comparable (133/273 vs. 48/108, Fig. 7A). However, further analysis showed that the percentage of DEGs in differentially methylated CRPC module genes was far higher than in differentially methylated primary PCa module (39/48 vs. 31/133, Fig. 7B).
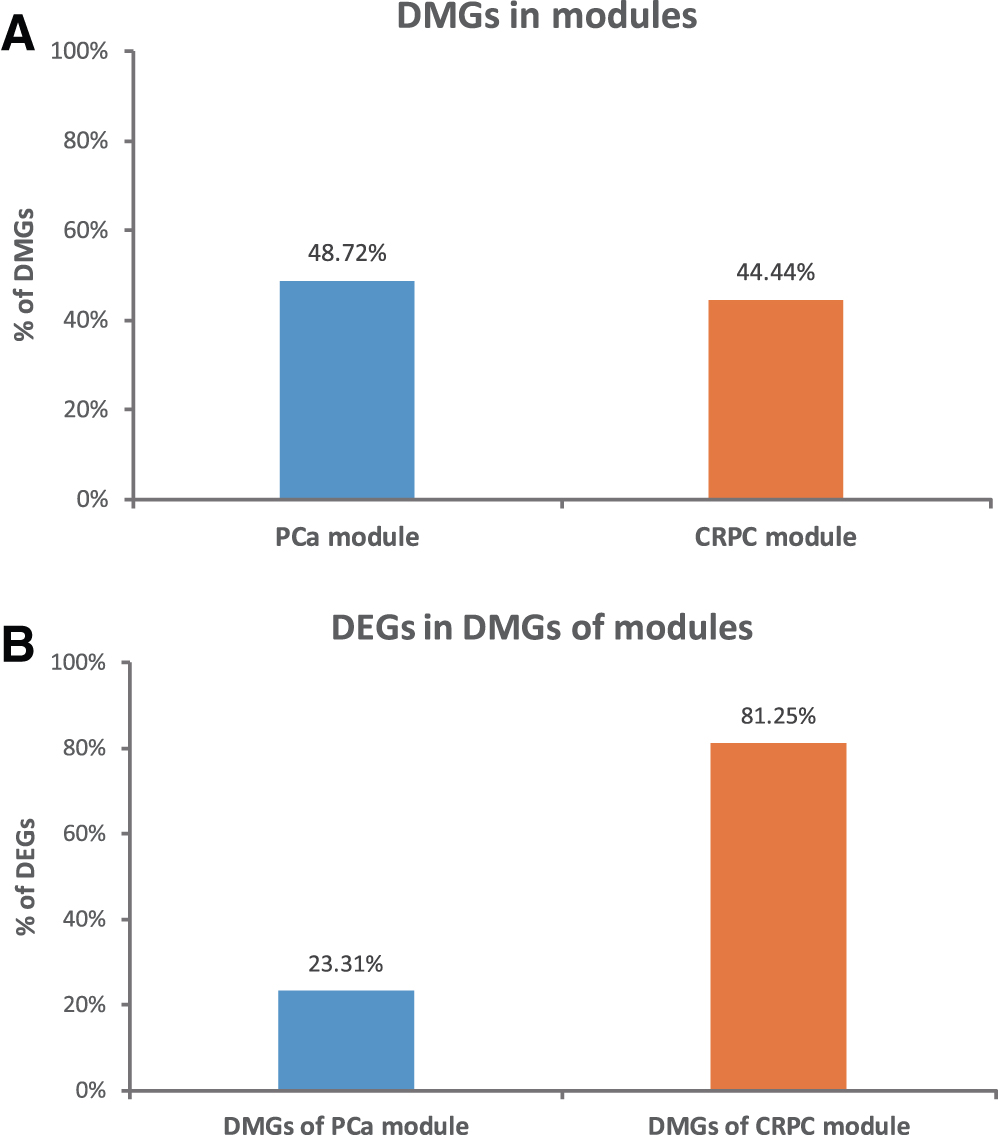
Identification of DMGs in functional modules.
Key TFs and ncRNAs regulating modules
The transcriptional regulation of associated genes is important in the progression of CRPC, and TFs could regulate the expression of these genes. The key TFs significantly regulating modules were predicted by pivot analysis based on TF-mRNA relationships. From 9396 TF-mRNA regulation relationships in the TRRUST V2 database, 33 key TFs regulating the primary PC and CRPC modules were identified through pivot analysis. Thirty-three total TF-module interactions were identified (p < 0.01) (Supplementary Table S9). Some significant key TFs are shown in Table 1.
Pivot (Transcription Factor)-Module Interactions
CRPC, castration-resistant prostate cancer; PCa, prostate cancer; TF, transcription factor.
ncRNAs also play important roles in gene regulation through many mechanisms. Similarly, pivot analysis was performed for key ncRNAs based on interactions between ncRNAs and genes. Searching 51,913 ncRNA-mRNA interaction relationships in the RAID 2.0 database, a total of 223 ncRNA-module interactions were identified in the primary PCa and CRPC modules (p < 0.01) (Supplementary Table S9). Some key ncRNAs are shown in Table 2.
Pivot (Noncoding RNA)-Module Interactions
ncRNA, noncoding RNA.
Prediction of potential drugs for primary PCa and CRPC
To explore therapeutic drugs for primary PCa and CRPC, we predicted the potential drugs of pathogenic genes based on relationships between drugs and target genes from the DrugBank database. First, we searched for known drugs for PCa. Four potential drugs specifically regulating modules in primary PCa and one drug specifically regulating modules in CRPC were identified (Table 3, connection ≥2, p < 0.05). When we expanded the analysis to all drugs in the database, there were 278 and 74 drugs regulating modules in primary PCa and CRPC, respectively (connection ≥2, p < 0.01) (Supplementary Table S10). Among these drugs, a total of 37 drugs could regulate modules in both groups. Thirty-seven drugs were identified to specifically regulate modules in CRPC. Among those specific CRPC module-targeted drugs, some drugs belong to phosphodiesterase-4 (PDE4) inhibitors, such as cilomilast, iloprost, piclamilast, rolipram, and roflumilast.
Pivot (Drug)-Module Interactions
Discussion
CRPC is an inevitable stage for most patients who receive ADT, even for some patients with radical therapy. In recent decades, many mechanisms and dysregulated genes that contribute to CRPC progression have been identified, but no consistent theory has been widely accepted. For CRPC, most drugs, even the new drugs apalutamide and darolutamide, other than chemotherapy and immunotherapy, still focus on targeting the androgen receptor (AR) signaling pathway. With the development of microarrays, next-generation sequencing and bioinformatics methods, an increasing number of genes have been identified to be related to CRPC. However, the global regulation of genes and signaling pathways in CRPC is still elusive. In this study, we constructed a PPI network based on DEGs and related genes, identified significant functional modules and their functions and pathways in primary PCa and CRPC, explored DMGs, key TFs, and ncRNAs regulating the modules, and predicted potential drugs for CRPC.
GSE32269 was downloaded from the GEO database, and differential expression analysis identified 588 DEGs between primary PCa and CRPC. To ensure that other genes related to PCa were not overlooked and to avoid the bias of only one selected GEO dataset, we also downloaded PCa-related gene lists from the NCBI-gene and OMIM databases for combined analysis. After constructing the PPI network and performing functional module analysis, we identified 22 and 14 significant modules in primary PCa and CRPC, respectively.
Centered on these modules, we performed GO functional and KEGG pathway analyses and identified distinct functions and pathways enriched in primary PCa and CRPC. For the KEGG pathway analysis, in primary PCa, the neuroactive ligand-receptor interaction, chemokine signaling pathway, and cytokine-cytokine receptor interaction were the top three pathways, while in CRPC, the cell cycle, protein digestion and absorption, and the calcium signaling pathway were the top three pathways. Moreover, further GO functional and KEGG pathway analyses for the CRPC-specific module genes also showed that DNA replication was enriched. This finding may imply that CRPC has molecular mechanisms distinct from those of primary PCa.
In the functional modules, some hub genes were identified in primary PCa and CRPC. Among these genes, LPAR1, LPAR3, and ANXA1 were also identified as hub genes in high-grade PCa in another study, although different GEO datasets were used (Foj and Filella, 2018). LPAR1 could promote epithelial maturation and block the invasion of PCa cells in three-dimensional culture (Härmä et al., 2012). SAA1 has been reported to be overexpressed in PCa tissue compared with normal tissue and plays a vital role in the fatty acid oxidation and cell growth promoted by Sun2 loss in PCa (Yajun et al., 2017). CASR may play a role in promoting PCa to metastasize to bone, and its expression was associated with an increased risk of lethal PCa (Ahearn et al., 2016).
Among the hub genes related to CRPC, PLK1 is a critical regulator of many cell cycle events and is significantly elevated upon the castration of mice carrying xenograft prostate tumors. Targeting PLK1 could enhance the efficacy of olaparib in CRPC (Li et al., 2017). CDC20 overexpression could facilitate the docetaxel resistance of a CRPC cell line (Wu et al., 2018). CDK1 could drive androgen-independent AR activity through AR S81 phosphorylation (Liu et al., 2017). It has been reported that the overexpression of BUB1B contributes to the growth and progression of PCa, and the survival analysis showed that BUB1B was an independent predictor of shorter biochemical recurrence-free survival (Fu et al., 2016). In this study, the expressions of CRPC modules hub genes PLK1, CDC20, CDK1, and BUB1B were verified in clinical samples of primary PCa and CRPC through RT-qPCR. The mRNA levels were consistent with the GEO microarray data. The mean expression of PLK1 was a little higher in CRPC samples than in primary PCa samples, although the difference was not significant. More samples may be needed to clarify its differential expression.
DNA methylation is one of the essential epigenetic mechanisms for gene expression (Damaschke et al., 2013). It involves the addition of a methyl group to the 5′-carbon of cytosine in CpG dinucleotide sequences. The methylation of CpG islands in promoter regions of genes may prevent or downregulate the gene transcription. Disturbances of DNA methylation could contribute to prostate tumorigenesis (Nowacka-Zawisza and Wiśnik, 2017). In this study, we analyzed the genome-wide DNA methylation in primary PCa and CRPC from GEO database, and DMGs in primary PCa and CRPC significant module genes were identified. We found that the states of DNA methylation in two groups were similar, but DMGs of CRPC module had more DEGs than DMGs of primary PCa. This may imply that the DNA methylation plays different roles in primary PCa and CRPC, and it would be more likely to cause differential gene expressions in CRPC.
In addition to DNA methylation, TFs and ncRNAs could also regulate gene expression. Through pivot analysis, we found 33 key TFs that regulate the modules of primary PCa and CRPC. Among them, E2F3, as one of the E2F TFs, could modulate EZH2 expression directly. The overexpression of the EZH2 gene has been implicated in the development of PCa (Foster et al., 2004). ZBTB16 is an androgen-induced tumor suppressor gene, and its low expression has been associated with a worse prognosis in PCa (Stopsack et al., 2019). MED1 was another key TF that regulates the CRPC modules. It is also an important coregulator that forms a transcription complex with the AR in PCa (Hsieh et al., 2014). ncRNAs are RNA transcripts that do not code proteins. However, ncRNAs could play important roles in cellular biological activities through multiple mechanisms, including development, differentiation, metabolism, X chromosome inactivation, genetic imprinting, chromatin modification, and transcription activation and inhibition (Sun et al., 2018). Transcription modulation is an important function of nuclear ncRNAs.
It has been reported that many lncRNAs are aberrantly expressed in PCa and may be new therapeutic targets of CRPC (Misawa et al., 2017). In this study, some key ncRNAs were identified by pivot analysis. Recently, studies have shown that TUG1 could promote PCa progression by regulating RLIM and DGCR8 and can predict a poor prognosis (Guo et al., 2019, Xu et al., 2019, Yang et al., 2019). MALAT1 is an ncRNA that contributes to castration resistance. MALAT1 was highly expressed in patients with a high Gleason score, prostate-specific antigen (PSA) level, and tumor stage, and in those with CRPC (Ren et al., 2013). Malat1 and AR-V7 were found to be increased in enzalutamide-resistant PCa cell lines. Targeting Malat1 with siRNAs could suppress enzalutamide resistance progression (Wang et al., 2017).
Based on the functional modules in primary PCa and CRPC, pivot analysis could help to predict potential drugs for PCa. Exisulind was identified as the only drug for the CRPC module among known drugs for PCa through pivot analysis. Exisulind is an orally active, apoptotic antineoplastic agent. As the sulfone derivative of sulindac, an NSAID, exisulind acts by inhibiting the enzyme cyclic guanosine monophosphate phosphodiesterase-5. As monotherapy, phase I data indicated that PSA progression may slow down in patients with biochemical recurrence after radical prostatectomy (Webster and Leibovich, 2005). However, a prospective, controlled phase II study of neoadjuvant exisulind therapy before radical prostatectomy showed no significant effect of exisulind on the biomarkers of cell death between biopsy specimens and radical prostatectomy specimens. More studies should be considered to evaluate a higher dose or longer duration of exisulind to evaluate its role (Weight et al., 2012).
Enzalutamide has been approved by the U.S. Food and Drug Administration (FDA) for treating both nonmetastatic CRPC and metastatic CRPC (Hussain et al., 2018). However, in this study, enzalutamide was identified as a specific drug for primary PCa, not CRPC. This discrepancy may be because enzalutamide is still an inhibitor of the androgen axis, which is an essential target for primary PCa. This may explain why CRPC could also eventually become resistant to enzalutamide, although it could take effect when first used (Claessens et al., 2014).
When we expanded candidate drugs to all drugs in DrugBank, PDE4 inhibitors were identified as a class of drugs that may have potential effects for CRPC. PDEs can hydrolyze cAMP and cGMP and play a major role in cellular signaling. The PDE4 gene family has four isoforms: PDE4A, PDE4B, PDE4C, and PDE4D. In a human prostatic tissue microarray, PDE4D was overexpressed in prostate adenocarcinoma samples when compared with benign prostatic hyperplasia samples (Rahrmann et al., 2009). Currently, the FDA-approved PDE4 inhibitor roflumilast is used in combination therapy for chronic obstructive pulmonary disease (Rabe, 2011). Other PDE4D inhibitors, such as the first-generation drug rolipram and second-generation cilomilast, were also developed and tested for the treatment of respiratory disorders. The effects of PDE4D in cancer are not well understood, and only a few studies have examined the role of PDE4D and its inhibitors in cancer therapy. Studies have shown that PDE4D inhibitors lead to decreased signaling of the sonic hedgehog (SHH), AR, and MAPK pathways in vitro. In vivo, PDE4D inhibitors could decrease the wet weight of PCa xenografts and increase apoptosis compared with vehicle-treated controls (Powers et al., 2015). These results suggested that PDE4D inhibitors may be an effective option for CRPC treatment.
Conclusions
In this study, we explored molecular mechanisms and identified potential drugs for CRPC from a bioinformatics view based on a thorough analysis of the global regulation of genes. Centered on the significant functional modules identified from DEGs and related genes, we found distinct GO functions and KEGG pathways in CRPC and identified DMGs, key interacting TFs and ncRNAs. Moreover, exisulind and PDE4 inhibitors were identified as potential drugs for treating CRPC. The results of this study provide a theoretical basis for further study on the mechanisms of CRPC progression and help in the development of new targeted therapies for CRPC.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by National Natural Science Foundation of China (31501052), Health Technology Innovation Project of Jilin Province, China (2017J049), and Jilin University Bethune Project B (2015338).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
Supplementary Table S8
Supplementary Table S9
Supplementary Table S10
Supplementary Figure S1
Supplementary Figure S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.