
Abstract
Lung adenocarcinoma (LUAD) accounts for an increasing proportion of non–small-cell lung cancer and an increasing number of cancer-related deaths worldwide. However, few biomarkers are available for prognosis and patient stratification. In all eight datasets from the Oncomine and The Cancer Genome Atlas (TCGA) LUAD cohorts, solute carrier family 2 member 1 (SLC2A1) was significantly more highly expressed in LUAD tissue than in normal lung tissue. High SLC2A1 expression was also significantly (p < 0.05) associated with a poor prognosis in stage I, II, and III subgroups using the Kaplan–Meier plotter. In the National Cancer Center of China (NCC) cohort, SLC2A1 expression correlated significantly (p < 0.05) with several parameters, including sex, smoking history, tumor size, tumor differentiation, T stage, N stage, and pathologic TNM stage. Moreover, multivariate Cox regression indicated SLC2A1 to be an independent prognostic factor (p < 0.05) in both TCGA and NCC cohorts. Eleven hallmark pathways were significantly enriched (p < 0.01, false discovery rate <0.25) in the high-SLC2A1 expression group. SLC2A1 is a promising biomarker that can be used to predict the prognosis of LUAD.
Introduction
Lung cancer is now the leading cause of cancer-related death in both males and females, and approximately four-fifths of lung cancer patients have non–small-cell lung cancer (NSCLC) (Torre et al., 2015; Chen et al., 2016). In recent years, lung adenocarcinoma (LUAD) has accounted for an increasing proportion of NSCLC, posing a great challenge to global health (Mao et al., 2016). With the development of molecular genetics, several key events in tumorigenesis and the development of lung cancer have been identified, including both the activation of oncogenes and inactivation of tumor suppressor genes (Barr Kumarakulasinghe et al., 2015; Bol et al., 2015; Yuan et al., 2015; Kalemkerian et al., 2018; Lindeman et al., 2018; Rizvi et al., 2018). Through this deep insight, the emergence of targeted therapy and immunotherapy has brought new hope for treatments, including EGFR tyrosine kinase inhibitors (EGFR TKIs) (Mok et al., 2017; Soria et al., 2018), receptor tyrosine kinase (RTK) inhibitors (Liao et al., 2015; Steuer et al., 2015) and immune checkpoint blockade (Postow et al., 2015; Lee et al., 2017). However, due to great heterogeneity, only a portion of patients benefit from these treatments, and overall survival remains disappointing. Therefore, additional molecular biomarkers are urgently needed.
It has long been known that tumor cells can promote proliferation and survival by altering metabolic patterns (Griffin and Shockcor, 2004; DeBerardinis et al., 2008; Boroughs and DeBerardinis, 2015). One of the most famous features of tumor cells is the Warburg effect (Liberti and Locasale, 2016), which is characterized by increased glucose uptake and the fermentation of glucose to lactate. Although the benefits of this phenomenon to tumor cells are not completely understood, studies have shown that it is involved in several functions, including supporting the biosynthetic requirements of rapid proliferation, providing an advantage for cell growth in the tumor microenvironment, and conferring cellular signals to tumor cells (Liberti and Locasale, 2016). Solute carrier family 2 member 1 (SLC2A1) encodes a major glucose transporter in the mammalian blood–brain barrier and is a crucial mediator of the Warburg effect (Lopez-Serra et al., 2014; Ooi and Gomperts, 2015). Recent studies have revealed that SLC2A1 is overexpressed and promotes biological processes involved in several cancer types, including glioma, gastric cancer, colorectal cancer, and lung cancer (Yan et al., 2015; Wang et al., 2017; Yang et al., 2017; Shi et al., 2019). Nonetheless, its potential as a biomarker in LUAD has not been carefully evaluated.
In general, the development of high-throughput sequencing has profoundly altered our understanding of tumorigenesis, metastasis, and chemoresistance (Stark et al., 2019). Moreover, a large number of public databases are available for exploration. Indeed, several biomarkers have been identified through a data mining approach and validated in independent cohorts. The Cancer Genome Atlas (TCGA,
The current study aimed to explore the prognostic value of the major glucose transporter SLC2A1 in LUAD using both public databases and an independent cohort from our hospital. The expression level of SLC2A1 between tumor and paired normal tissues was analyzed first, after which its association between SLC2A1 expression and clinicopathological parameters was assessed. Kaplan–Meier (KM) curve and Cox multivariate regression analyses were also performed in the LUAD cohort of TCGA and our own cohort to evaluate the potential for SLC2A1 to predict prognosis. Finally, gene set enrichment analysis (GSEA) was conducted to identify SLC2A1-related pathways and determine potential targets for further study.
Materials and Methods
Patient samples
All clinical patient samples in this study were collected between June 2006 and June 2014 at the National Cancer Center/Cancer Hospital, CAMS. A total of 415 patients with LUAD who underwent R0 resection were included in the analysis. The entire enrollment process is shown in Figure 1. All patients provided informed consent before surgery. The collected clinical information included sex, age, tumor location, tumor differentiation status, T stage, lymph node metastasis, and TNM stage. All of the specimens were pathologically confirmed by two experienced pathologists and stored properly in our biobank. The 8th edition of the TNM staging system was used to guide pathological classification of the primary tumor and the degree of lymph node metastasis.
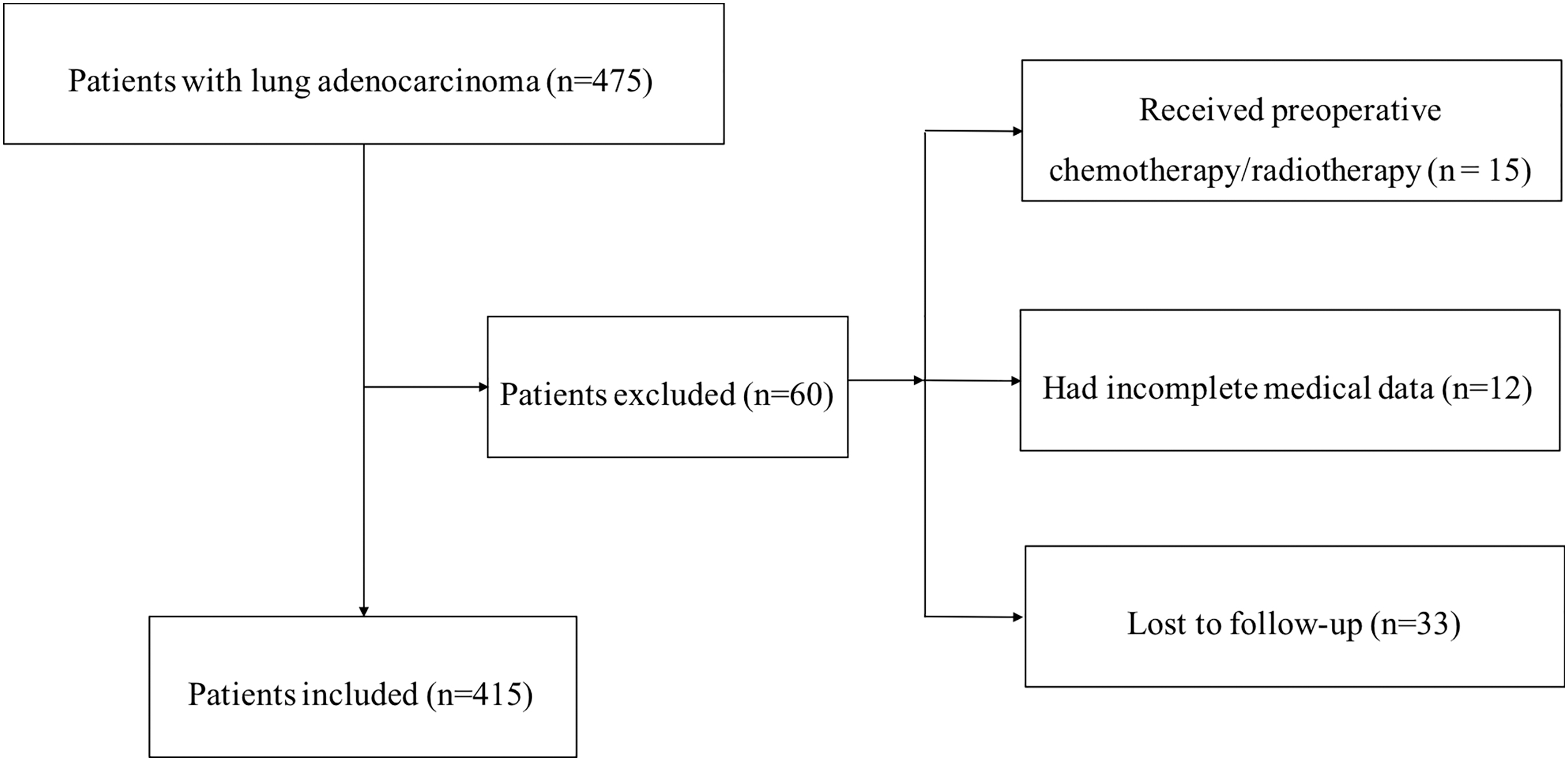
Flowchart of the enrollment process.
The current study was conducted in accordance with the Declaration of Helsinki. The Clinical Research Ethics Committee of the National Cancer Center/Cancer Hospital, CAMS approved this study (19/305-2089). Patients were followed up in the outpatient department every 3–6 months for the first 2 years after surgery and then annually until March 4, 2019. The follow-up material consisted of medical history, physical examinations, and chest computed tomography.
mRNA expression and clinical information in the dataset from TCGA
RNA-seq expression profiles and clinical information of the LUAD cohort of TCGA (535 patients) were downloaded from the Genomic Data Commons (GDC) portal (
SLC2A1 expression analysis using Oncomine and gene expression profiling interactive analysis
Oncomine is a web-based application containing more than 700 independent datasets with curated data and consistent oncological terms and interpretations (Rhodes et al., 2004). Eight LUAD datasets, including both tumor and normal tissues were extracted for SLC2A1 expression analysis. Gene expression profiling interactive analysis (GEPIA) is another newly developed website that uses RNA-seq data from TCGA and the Genotype-Tissue Expression (GTEx) project (Tang et al., 2017); it is commonly used to compare gene expression profiles between different cancer types and to perform survival analysis. In this study, we used both Oncomine and GEPIA to preliminarily explore SLC2A1 expression. In addition, survival analysis of the LUAD cohort of TCGA was conducted using GEPIA. The median TPM value was chosen as the cutoff point for grouping.
Survival analysis of SLC2A1 in the lung cancer database with the KM plotter
The KM plotter is an online survival analysis tool that integrates datasets from the Gene Expression Omnibus (GEO), European Genome-phenome Archive (EGA) and TCGA (Győrffy et al., 2013). It combines gene expression and clinical data and calculates both the hazard ratio (HR) and log-rank p value. In the present study, the KM plotter was used to explore the prognostic value of SLC2A1 expression in different cohorts and in subgroups stratified by TNM stage. The median expression value was used as the cutoff.
GSEA of SLC2A1 and function of the SLC2A1 network
To identify pathways closely related to SLC2A1, GSEA was performed on data for 515 LUAD patients from TCGA. GSEA is a powerful computational method that identifies a significantly aggregated biological function or molecular pathway between two different biological states (Subramanian et al., 2005). Patients were divided into groups of high and low expression based on the median expression level of SLC2A1. Hallmark gene sets containing 50 gene sets were employed for GSEA. According to the recommended parameters, the number of permutations was set to 1000, and the enrichment statistic was set to “weighted.” Pathways with p < 0.01 and FDR <0.25 were considered significant, as listed in the Results section.
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, version 11.0) online database (Franceschini et al., 2012) was employed to construct a protein–protein interaction (PPI) network centered on SLC2A1 and for functional annotation using Gene Ontology (GO) (Ashburner et al., 2000) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Kanehisa, 2002). An interaction score >0.4 was considered significant.
Tissue microarrays
Formalin-fixed paraffin-embedded (FFPE) samples from LUAD patients stained with Hematoxylin and Eosin (H&E) were reviewed independently by two experienced pathologists. A representative area, ∼4 μm thick, harboring a high tumor/stroma ratio was selected for each sample. In total, the tissue microarray (TMA) contained 415 LUAD cases from the National Cancer Hospital, CAMS.
Immunohistochemistry of SLC2A1
TMAs were first deparaffinized, rehydrated, and treated with 2 N HCl for 15 min, followed by 100 mM Tris-HCl (pH 8.5) for 10 min. The TMAs were then blocked with 3% H2O2 for 30 min and goat serum at room temperature for 30 min. After blocking, a rabbit anti-SLC2A1 polyclonal antibody (1:2000, HPA004117; Sigma-Aldrich, St. Louis, MO) was added and incubated at 4°C overnight, followed by a polyclonal peroxidase-conjugated anti-rabbit IgG (Zhongshanjinqiao, Beijing, China) at room temperature for 20 min according to the manufacturer's instructions.
Immunohistochemistry (IHC) staining was evaluated and scored based on the density and percentage of SLC2A1-positive tumor cells by two experienced pathologists who were blinded to the clinical information of the LUAD patients. An immunoreactive score for each case was calculated based on the product of the following multipliers: (1) scores representing staining intensity (no staining = 0, weak = 1, moderate = 2, strong = 3) and (2) scores representing the positive staining area (0–25% = 0, 25–50% = 1, >50% = 2). In the current study, tissues with scores ≤1 were grouped together and considered negative for SLC2A1; tissues with scores ≥2 were considered positive for SLC2A1.
Statistical analyses
Statistical analyses were performed by R version 3.5.3 (The R Foundation for Statistical Computing). Supplementary Table S1 of clinical data and SLC2A1 expression was analyzed using the χ2 test or Fisher's exact test. Survival analysis was performed using the KM method, and p values were calculated using the log-rank test. Associations between SLC2A1 expression values and clinicopathological characteristics were determined by the Kruskal–Wallis test and Wilcoxon test. Multivariate Cox regression was applied to identify independent prognostic variables, and the median TPM value was set as the cutoff. A p value <0.05 was considered statistically significant.
Results
Expression analysis of SLC2A1 in LUAD
SLC2A1 expression analysis was first conducted in various cancer types using the Oncomine database. The results showed SLC2A1 to be significantly highly expressed in several tumor types, including bladder cancer, breast cancer, colorectal cancer, esophageal cancer, gastric cancer, head and neck cancer, kidney cancer, leukemia, lung cancer, lymphoma, ovarian cancer, pancreatic cancer, and many other cancers (Fig. 2A). TCGA pan-cancer SLC2A1 expression analysis using GEPIA revealed similar results (i.e., SLC2A1 expression was elevated in multiple cancer types) (Fig. 2B). SLC2A1 expression was significantly higher in tumor tissues than in normal tissues in all datasets (Table 1). Significantly upregulated SLC2A1 expression was also found in cancerous tissue in the LUAD cohort from TCGA (Fig. 2C). In addition, the KM curve showed that the prognosis of LUAD patients with high SLC2A1 expression was significantly worse than that of LUAD patients with low SLC2A1 expression (Fig. 2D).
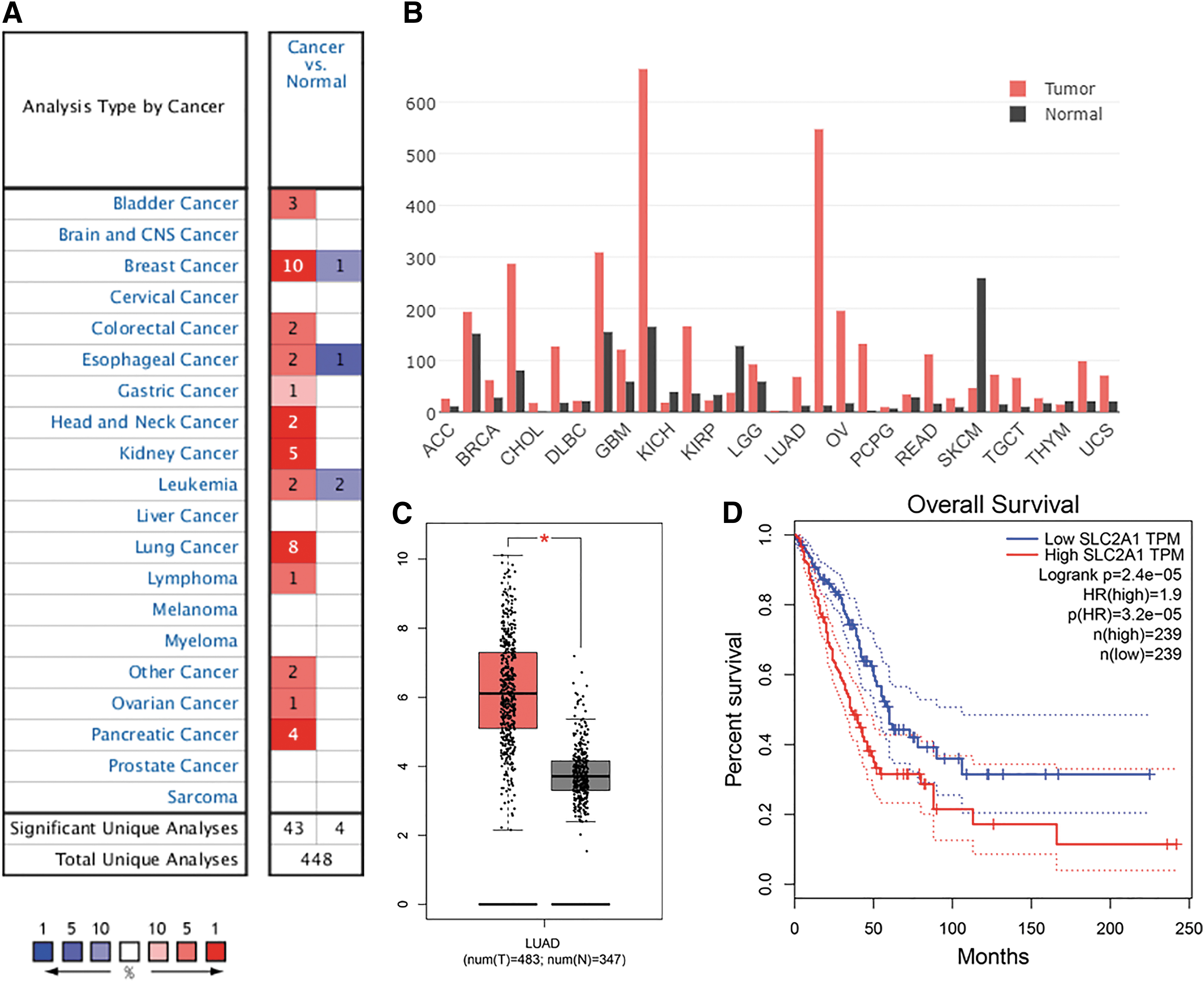
Aberrant SLC2A1 expression and prognostic value in cancer.
The Significant Changes of SLC2A1 Expression in Transcription Level Between Lung Adenocarcinoma and Normal Tissues
Source: Oncomine database.
SLC2A1, solute carrier family 2 member 1.
Association between SLC2A1 expression and clinicopathological factors in the cohort from TCGA
Comparison of SLC2A1 expression grouped by clinical indicators revealed consistent results based on TCGA LUAD data. The clinicopathological factors that were extracted for analysis included age, sex, race, smoking history, N stage, and pathologic stage. In multiple comparisons, all six tests showed great significance (p < 2.2e-16) (Fig. 3A–F). Furthermore, after stratification by different clinical factors, significant differences were observed between each tumor subgroup and the reference/normal group (p < 0.05) (Fig. 3A–F). Therefore, to further validate the prognostic value of SLC2A1, multivariate Cox regression analysis (Table 2) was performed, and the results illustrated that T stage (HR: 1.310, 95% confidence interval [CI]: 1.079–1.591), N stage (HR: 2.319, 95% CI: 1.711–3.142), and SLC2A1 expression (HR: 1.152, 95% CI: 1.016–1.306) were independent prognostic factors (p < 0.05) for the LUAD cohort from TCGA. In contrast, M stage was not an independent prognostic factor in the multivariate Cox model.

The relationship between SLC2A1 expression and clinical parameters (TCGA). Association between SLC2A1 relative expression and
Multivariate Cox Proportional Hazards Regression on Lung Adenocarcinoma Overall Survival
Source: The Cancer Genome Atlas.
CI, confidence interval; HR, hazard ratio.
Prognostic analysis of SLC2A1 using the KM plotter
A total of 720 LUAD patients with SLC2A1 probe information were included in analysis using KM plotter. The KM curve containing all patients demonstrated that high SLC2A1 expression was significantly (p < 1.2e-10) (Fig. 4A) associated with poor overall survival, and the role of SLC2A1 in prognosis was consistent after the patients were stratified into different subgroups based on TNM stage and N stage. High SLC2A1 expression was significantly (p < 0.05) associated with a poor prognosis in the stage I, stage II, stage III, N0, and N1 subgroups (Fig. 4B–F).

SLC2A1 expression analysis and survival analysis of LUAD patients in KM-Plot lung cancer database.
Correlations between SLC2A1 expression and clinicopathological parameters in the National Cancer Center of China cohort
As shown in Table 3, χ2 tests were employed to assess correlation between SLC2A1 expression (high and low) and clinicopathological parameters, and the results revealed SLC2A1 expression to be significantly associated with sex, smoking history, tumor size, tumor differentiation, T stage, N stage, pathologic TNM stage, and therapeutic strategy. It can also be seen from the table that patients with high levels of SLC2A1 expression tended to present at advanced stages, which suggests that SLC2A1 is linked to patient prognosis.
Correlations Between SLC2A Expression and Clinicopathological Parameters of 415 Patients with Lung Adenocarcinoma
Boldface indicates p value < 0.05.
Validation of the prognostic value of SLC2A1 in the NCC cohort
A total of 415 LUAD patients from our NCC cohort were divided into high (n = 151) and low SLC2A1 expression groups according to IHC staining results. Representative photomicrographs of H&E and SLC2A1 immunohistochemical staining are shown in Figure 5. A KM curve was generated using all patients and subgroups. The results first revealed that high SLC2A1 expression was significantly (p < 0.001) (Fig. 6A) associated with poor overall survival. In subgroup analysis, SLC2A1 remained a significant indicator (p < 0.05) of a poor prognosis in stage I, stage II, stage III, N0, and N+ (patients with lymphatic metastasis) subgroups (Fig. 6B–F). To further verify its independent prognostic value, univariate and multivariate Cox regression analyses were performed (Table 4). In the univariate analysis, overall survival was significantly associated with age, sex, smoking history, tumor size, tumor differentiation, T stage, lymph node metastasis, pathologic TNM stage, therapeutic strategy, and SLC2A1 expression (p < 0.05). In the subsequent multivariate analysis, factors, including patient age, tumor size, lymph node metastasis, therapeutic strategy, and SLC2A1 expression remained significantly associated with overall survival.

Representative photomicrographs of LUAD TMA sections.

Kaplan–Meier curves showing survival of the 415 patients with LUAD according to SUSD2 expression.
Univariate Analysis and Multivariate Analysis of Risk Factors for Prognosis of 415 Lung Adenocarcinoma Patients
Boldface indicates p value < 0.05.
GSEA and functional annotation of the SLC2A1 PPI network
A PPI network was constructed using 10 genes identified from the STRING database significantly associated with SLC2A1, namely, STOM, GIPC1, LDHA, HIF1A, AKT1, TP53, SMUG1, HK2, VEGFA, and INS (Fig. 7A). All of these genes retained an interactive score >0.4. To investigate the potential role of this network, functional annotation analyses (GO and KEGG) were carried out, and the significant enrichment terms are shown in Figure 7B. To investigate the biological processes in which SLC2A1 may be involved, GSEA was performed, and the results showed glycolysis, the G2M checkpoint, MTORC1 signaling, and hypoxia to be the most significantly associated processes (Fig. 8A–D and Table 5). Detailed information on each biological process is listed in Tables 6 –9. The top 100 genes that were significantly positive or negatively correlated with SLC2A1 are displayed in Figure 8E.
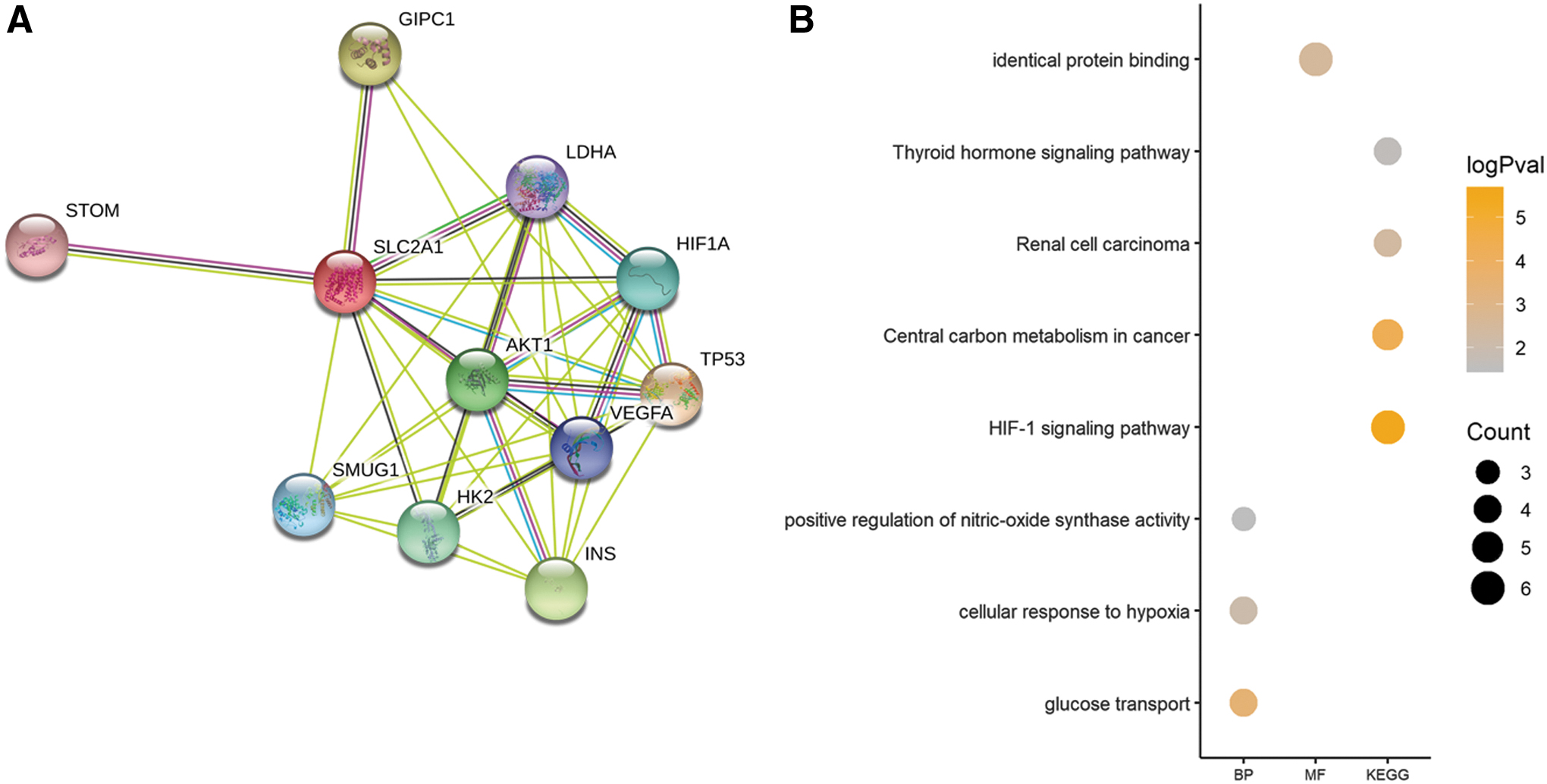
PPI network and functional annotation.
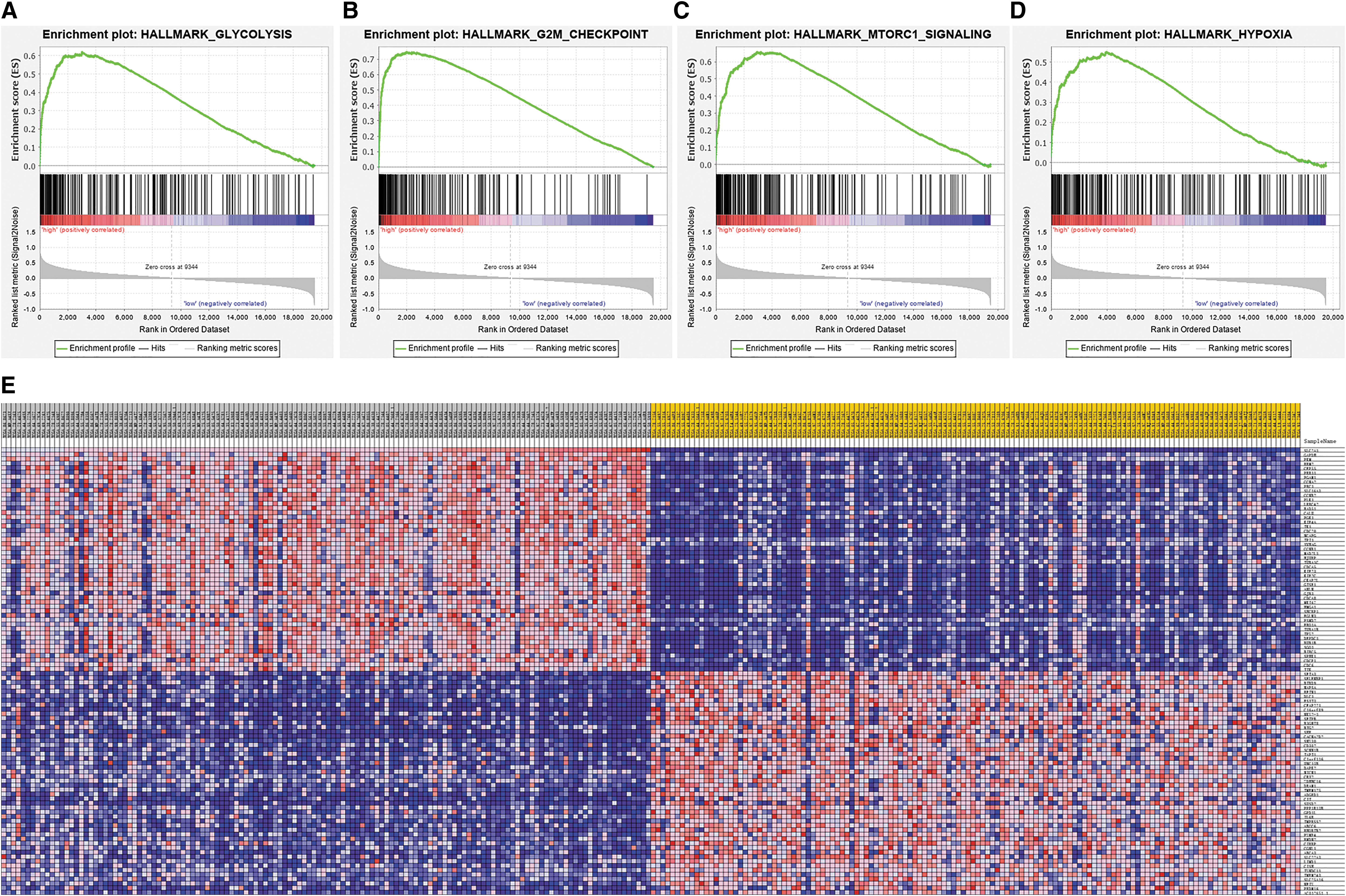
Gene Set Enrichment Analysis centered on SLC2A1.
Top Biological Processes Enriched in Lung Adenocarcinoma Centered on SLC2A1
ES, enrichment score; NES, normalized enrichment score; NOM, nominal p-value; FDR, false discovery rate; FWER, familywise-error rate.
Gene Set Enrichment Analysis Detail of Hallmark Glycolysis Pathway
ES, enrichment score; Running ES is the enrichment score for this set at this point in the ranked list of genes.
Gene Set Enrichment Analysis Detail of Hallmark G2M Checkpoint
Gene Set Enrichment Analysis Detail of Hallmark MTORC1 Signaling
Gene Set Enrichment Analysis Detail of Hallmark Hypoxia Pathway
Discussion
LUAD is a highly malignant and heterogeneous disease with various prognoses. Patients at the same stage may have quite different outcomes, even at an early stage. Thus, it is urgent to identify novel biomarkers to fine-tune the prediction of patient prognosis and to adjust treatment strategies. In the present study, we integrated mRNA expression with immunohistochemical staining analyses to investigate the significance, prognostic value, and hypothetical mechanism of SLC2A1 in LUAD.
SLC2A1 has been widely studied as the primary glucose transporter, and it has been identified as a probable prognostic factor in several cancer types. For example, Huang and colleagues performed integrated bioinformatics analysis and revealed the potential research value of the SLC family in small cell lung cancer (Liao et al., 2019). Concerning NSCLC, a meta-analysis, including 26 studies, was conducted in 2018, and the results suggested that positive SLC2A1 expression predicted a poor prognosis (Zhang et al., 2018). Nonetheless, there are several problems associated with previous studies, including the ambiguous cutoff value of SLC2A1 expression between different studies and incomplete prognostic information in some studies. In addition, conflicting results have been obtained in different studies when multivariate analysis was used. Another study explored the association of single nucleotide polymorphisms of SLC2A1 and patient prognosis and demonstrated that SLC2A1 variants may be useful in predicting the survival of those with early stage NSCLC (Do et al., 2018). Gao and Wang (2018) also conducted an in silico study and revealed SLC2A1 as a prognosis-related differentially expressed gene in LUAD; however, only two datasets were included, and the survival analysis was presented using the KM curve and did not exclude the influence of other factors. Thus, the prognostic value of SLC2A1 in LUAD needs to be verified.
In our study, the cohort from TCGA and eight independent datasets consistently showed that SLC2A1 was significantly highly expressed in LUAD tissues compared with paired normal tissues, indicating that SLC2A1 plays a role in tumorigenesis and progression. Furthermore, we demonstrated that SLC2A1 has great potential in predicting patient prognosis. High SLC2A1 expression correlated significantly with poor overall survival in multiple datasets, including those from TCGA, the KM plotter, and our own cohort. Additionally, subgroup analysis indicated that SLC2A1 expression can be used to stratify the prognosis of patients with early stage LUAD. It may also be useful when identifying early stage patients with undesirable prognoses and subsequently guiding their therapeutic regimen. SLC2A1 was further shown to be an independent factor in both the dataset from TCGA and our dataset, indicating its potential application as a complementary marker for the TNM staging system. It should be noted that investigating whether SLC2A1 may be used as a diagnostic marker is beyond the scope of this study.
With respect to the molecular mechanism involved, an in vitro study conducted by Wang et al. (2017) showed that knockdown of SLC2A1 in LUAD cells inhibits cellular proliferation, metastasis, and glucose utilization. High SLC2A1 expression is significantly associated with the cell cycle pathway. Another study confirmed these results and demonstrated that suppressing SLC2A1 may lead to diminished tumor growth in vivo (Ooi and Gomperts, 2015). Furthermore, an SLC2A1 inhibitor exhibited antitumor activity, even in chemoresistant cancer cells (Ooi and Gomperts, 2015). Our results are highly similar to those from previous pathway analyses, and we utilized more samples for exploration and identified 14 enriched hallmark pathways, including the G2M checkpoint, hypoxia, and MTORC1 signaling. These specific pathways need to be further verified both in vitro and in vivo.
There are certain limitations to the present study. First, the immunohistochemical cohort from our hospital was obtained from a single center, and the data were collected retrospectively. Thus, bias is inevitable. Second, all of our samples were obtained by surgical resection. Therefore, the molecular biology characteristics may not be extrapolated to the entire LUAD cohort. Prospective studies using multicenter cohorts are needed to verify these results. In summary, this is the first time in which in silico analysis was combined with a large cohort to immunohistochemically analyze and verify the relationship between SLC2A1 and LUAD. Our results strongly support the prognostic value of SLC2A1 in LUAD.
Footnotes
Acknowledgment
The authors thank all staff in the Department of Thoracic Surgery for their support during the study.
Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Key R&D Program of China (2017YFC1311000, 2018YFC1312100), CAMS Initiative for Innovative Medicine (2017-I2M-1-005, 2017-I2M-2-003, 2019-I2M-2-002), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2018PT32033,2017PT32017), Innovation team development project of Ministry of Education (IRT_17R10).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.