
Abstract
Mitochondria serve as a hub for cell metabolism, energy production, and reactive oxygen species generation through multiple signaling pathways that control cardiomyocyte survival and death and contribute to the pathophysiological process of cardiovascular diseases (CVDs). Therefore, maintaining mitochondrial quality control (MQC) is essential for mitochondrial homeostasis and cardiac health. The Hippo pathway is a conserved signaling cascade that regulates mitochondrial function and cardiomyocyte fate and plays a crucial role in cardiac regeneration, repair, and diseases. MQC and the Hippo pathway are tightly coupled in CVDs, and several regulatory elements of the Hippo pathway directly/indirectly regulate mitochondria-related proteins to mediate mitochondrial morphology and function. In turn, mitochondrial morphology partly influences Hippo pathway function in the heart. This review outlines the underlying mechanisms by which the Hippo pathway regulates MQC from a variety of perspectives, including mitochondrial dynamics, mitophagy, mitochondrial biogenesis, mitochondrial apoptosis under oxidative stress, and mitochondrial metabolism. We also discuss the roles of the Hippo pathway in orchestrating mitochondria in CVDs, such as myocardial ischemia–reperfusion injury, septic cardiomyopathy, and diabetic cardiomyopathy, which may provide a novel therapeutic strategy.
Introduction
In mammals, compared with other organs, the heart is especially vulnerable to organ failure owing to its poor capacity for self-renewal. Investigations in the field of myocardial regeneration and heart repair are still in the nascent stage. In the past decade, the Hippo pathway has attracted attention because of its highly conserved kinase chain, which plays a key role in cardiovascular diseases (CVDs) (Wang et al., 2016, 2018b; Leach et al., 2017; Morikawa et al., 2017; Yu et al., 2019b; Shang et al., 2020a).
The Hippo pathway, which is found both in Drosophila and mammals, contains a core kinase cassette that includes mammalian STE20-like protein kinases 1 and 2 (MST1/2; homologs of Hpo in Drosophila melanogaster), large tumor suppressor kinases 1 and 2 (LATS1/2; Warts homologs), and their respective adaptor proteins, Salvador 1 (SAV1; a homolog of SAV), MOB kinase activators 1A and 1B (MOB1A/1B; homologs of Mats), and the downstream effectors of the Hippo signaling pathway-transcriptional coactivators YAP (yes-associated protein) and TAZ (transcriptional coactivator with PDZ-binding motif; the two homologs of Yorkie; YAP, encoded by YAP1; TAZ, also known as WWTR1) (Wang et al., 2018a; Mia and Singh, 2019; Moya and Halder, 2019).
Upstream signaling pathways of the Hippo pathway, such as the oxidative stress response, pressure overload, and mechanical signaling, directly inactivate the Hippo pathway after cardiac injury (Nakamura et al., 2016; Wang et al., 2016; Byun et al., 2019). Once the Hippo pathway is “ON,” activated MST1/2 recruits SAV1 and MOB1A/B to assist MST1/2 in the phosphorylation of LATS1/2. In turn, activated LATS1/2 promotes YAP and TAZ phosphorylation. Activated YAP binds the scaffolding molecule 14-3-3 in the cytoplasm, leading to cytoplasmic sequestration and inhibition of transcription coactivator function, with eventual shuttling to the proteasome for further ubiquitylation–degradation.
When Hippo signaling is “OFF,” dephosphorylated YAP partners with TAZ, localizes to the nucleus, binds different transcription cofactors, and is subsequently directly involved in cardiomyocyte survival and death through targeting of downstream genes related to regeneration, antiapoptosis, and antioxidant scavengers (Nakamura et al., 2016; Morikawa et al., 2017; Wang et al., 2018a) (Fig. 1). This process is partly and simultaneously regulated by the Hippo pathway–mitochondria interaction.

The Hippo pathway in mammals. When the Hippo pathway is “ON,” triggering a phosphorylation cascade reaction, YAP is retained in the cytoplasm and degraded. When the Hippo pathway is “OFF,” YAP translocates into the nucleus and impacts target genes. LATS1/2, large tumor suppressor kinases 1 and 2; MOB1A/1B, MOB kinase activators 1A and 1B; MST1/2, mammalian STE20-like protein kinases 1 and 2; SAV1, Salvador 1; TAZ, transcriptional coactivator with PDZ-binding motif; YAP, yes-associated protein. Color images are available online.
Mitochondria are “power packs” for the heart, supplying ATP by oxidative phosphorylation (OXPHOS), producing reactive oxygen species (ROS), and utilizing and biosynthesizing various biomolecules for tissue growth and cell proliferation. Mitochondria undergo continuous and dynamic changes to maintain homeostasis of morphology, number, and quality, which is known as mitochondrial quality control (MQC). MQC includes mitochondrial dynamics (mitochondrial fission and fusion), mitophagy, mitochondrial biogenesis, mitochondrial apoptosis, mitochondrial redox and energetics balance, and mitochondrial proteostasis; these are critical mechanisms with important roles in physiology and disease in the heart (Murphy et al., 2016).
MQC determines cardiomyocyte survival or death by integrating multiple cellular signaling pathways, including the Hippo pathway (Arrazola et al., 2015; Nakamura et al., 2016; Zhang et al., 2017a; Wang et al., 2018a; Shang et al., 2020a). In both Drosophila and mammals, YAP/Yorkie (the downstream effector of the Hippo pathway) is initiated by upstream stimuli, such as oxidative stress, and directly regulates a series of mitochondrial genes to control cardiac cell fate and play critical roles in various heart diseases (Wang et al., 2019; Yu et al., 2019a, 2019b).
In addition, other components of the Hippo pathway have a direct or indirect effect on mitochondria-related proteins (Del Re et al., 2014; Feng et al., 2018; Tian et al., 2019). Concurrently, some mitochondrial dynamics-related proteins also act on the Hippo pathway to execute its function (Deng et al., 2016). Overall, the Hippo pathway regulates mitochondrial morphology and function in various ways, and mitochondrial morphology partly influences the role of the Hippo pathway in the pathological process of CVDs.
In this review, we summarize the current knowledge regarding the mechanisms that underlie the Hippo pathway and regulate mitochondrial structure and function through mitochondrial dynamics, mitophagy, mitochondrial apoptosis, and mitochondrial metabolism. Furthermore, we discuss how the Hippo pathway orchestrates MQC and plays a role in the pathological process of myocardial ischemia–reperfusion injury (MI/RI), septic cardiomyopathy and diabetic cardiomyopathy (DCM).
Cross-Talk Between the Hippo Pathway and Mitochondrial Dynamics
In most cells, a hyper-interconnective network is formed between mitochondria through continuing mitochondrial fusion and fission, which is also called mitochondrial dynamics. The balance of mitochondrial dynamics is tightly regulated by the interactions of various fission and fusion proteins situated in the mitochondrial double membrane. In mammalian cells, mitochondrial fusion is mainly controlled by mitofusin1 and 2 (Mfn1/2), and optic atrophy protein 1 (OPA1), whereas mitochondrial fission is modulated by dynamin-related protein 1 (Drp1), the mitochondrial fission 1 (Fis1) protein, and mitochondrial fission factor (MFF).
When mitochondria are damaged, the injured mitochondria divide into two daughter mitochondria, the depolarized and unhealthy mitochondria are removed, and the healthy mitochondria are further fused and repaired. Mitochondrial dynamics maintain the integrity of the mitochondria to protect cardiomyocytes and favor physiological function of the heart, whereas the imbalance of mitochondrial fission and fusion causes various CVDs through accumulation of damaged mitochondria (Tahrir et al., 2019).
Accumulating evidence demonstrates that mitochondrial dynamics-related proteins are an important target of the Hippo pathway in both flies and mammalian cells (Nagaraj et al., 2012; Deng et al., 2016) (Fig. 2). In both Drosophila and human cell lines, genome-wide microarray studies showed an increase in mitochondrial expansion through the activation of Yki or YAP2 through direct targeting of several mitochondria-related genes, including mitochondrial fusion genes—OPA1 and mitochondrial assembly regulatory factor (Marf, a homolog of MNF1 and 2 in humans). Deletion of Opa1 and Marf can suppress Yki-mediated tissue growth (Nagaraj et al., 2012).

The Hippo pathway and mitochondrial dynamics, mitophagy, and mitochondrial biogenesis. When Hippo signaling is “off,” YAP combines with TAZ, translational coactivator with PDZ-binding motif, and TEAD, TEA domain, family members to form complexes that directly or indirectly target mitochondria-related genes to regulate mitochondrial dynamics, mitophagy, and mitochondrial biogenesis. The mitochondria-related genes in turn partly react to the Hippo pathway. Arrows or blunt ends indicate activation or inhibition, respectively. Mitochondrial dynamics-related genes: ChChd3, coiled-coil-helix-coiled-coil-helix domain-containing 3; Drp1, dynamin-related protein 1; Mfn1/2, mitofusin1/2; OPA1, optic atrophy protein 1. Mitophagy-related genes: Bnip3, Bcl-2/adenovirus E1B 19 kDa interacting protein 3; FUNDC1, FUN14 domain-containing protein 1; Parkin, encoded by PARK2. Mitochondrial biogenesis-related genes: ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; PGC1-α, peroxisome proliferator-activated receptor-α coactivator 1. Color images are available online.
Subsequent findings identified another novel mitochondrial fusion gene, coiled-coil-helix-coiled-coil-helix domain-containing 3 (ChChd3), which is localized in the inner mitochondrial membrane (IMM) and is targeted by the Hippo pathway in Drosophila and mammalian cells (Deng et al., 2016). The loss of ChChd3 can partially induce tissue undergrowth by inactivating the Hippo pathway (Deng et al., 2016). These results indicate that there may be a bidirectional relationship between mitochondrial fusion and the Hippo pathway, which may be important in organ development and disease pathogenesis. This cross-talk between the Hippo/YAP pathway and mitochondrial fusion partly promotes mitochondrial renewal to compensate for the decrease in cell proliferation owing to defective mitochondrial fusion.
Several studies have shown that the Hippo pathway regulates OPA1-related mitochondrial fusion and plays an essential role in ischemia/reperfusion (I/R) injury (Feng et al., 2018; Ma and Dong, 2019; Wei et al., 2019).
I/R injury induces mitochondrial dysfunction, whereas enhanced mitochondrial fusion can improve organ perfusion by promoting mitochondrial communication and repair. Ma and Dong (2019) illustrated that hypoxia-induced suppression of OPA1-related mitochondrial fusion through the YAP-Hippo pathway is involved in the pathogenesis of MI/RI in vivo and in vitro. However, whether other elements of the Hippo pathway mediate mitochondrial fusion and contribute to the processes of cardiac pathology remains to be clarified. In addition, OPA1 includes two isoforms, long OPA1 (L-OPA1) and short OPA1 (S-OPA1); L-OPA1 mediates mitochondrial fusion and is then cleaved into S-OPA1. Which isoform is directly regulated by YAP is still poorly understood.
Although the Hippo-YAP pathway directly targets diverse mitochondria-associated genes, the transcription of mitochondrial fission-related genes is not directly targeted by YAP (Wang and Song, 2018; Shang et al., 2019; Tan et al., 2019; Yu et al., 2019a). Drp1 activation and translocation from the cytoplasm to the OMM is a prerequisite for mitochondrial fission. c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), AMPK, and DUSP1 have been found to be involved in signaling upstream of the mitochondrial translocation of Drp1 (Wang and Song, 2018).
In different CVDs, the Hippo pathway has been verified to regulate Drp1-related mitochondrial fission through molecular signaling. After myocardial infarction, Mst1 KO mouse heart tissue fibrosis and myocardial cell death were inhibited because of decreased mitochondrial fission. At the molecular level, Mst1 gene knockout may reduce excessive mitochondrial fission through the JNK-Drp1 pathway (Wang and Song, 2018). Yu et al. demonstrated that in Tg YAP (YAP transgenic) mice with septic cardiomyopathy, the overexpression of YAP attenuates lipopolysaccharide (LPS)-induced myocardial injury and heart dysfunction.
In vitro, YAP overexpression improves myocardial cell survival by inhibiting Drp1-related mitochondrial fission through the mitogen-activated protein kinase (MAPK)-ERK signaling pathway (Yu et al., 2019a). Tan et al. (2019) found that irisin treatment attenuated myocardial death and cardiac dysfunction in mice with septic cardiomyopathy, and this process was caused by suppression of Drp1-related mitochondrial fission through downregulation of the JNK-LATS2 signaling pathway in vitro. In addition, a study revealed that in mice with septic cardiomyopathy, the expression of Mst1 is upregulated, Mst1 ablation results in suppression of Drp1-induced mitochondrial fission, and F-actin is one of the downstream molecules of mitochondrial fission (Shang et al., 2019).
These results implied that the Hippo pathway regulates mitochondrial fission indirectly, but downstream mitochondrial fission needs more exploration. In addition, it is unclear whether fragmented mitochondria are indeed harmful, especially in cardiomyocytes wherein mitochondria are fragmented (Bhandari et al., 2015). During healthy and unhealthy mitochondrial interexchange, mitochondrial fusion causes “pollution” of healthy mitochondria, whereas mitochondrial fission also helps the damaged mitochondria by removing harmful content (Shirihai et al., 2015).
Cross-talk between the Hippo-YAP pathway and mitochondrial fission has also been observed (von Eyss et al., 2015). In mammary epithelial cells and breast cancer, MYC inhibits the translocation of YAP/TAZ in a manner dependent on the activated AMPK pathway, and this process can be inhibited by a decrease in Drp1-mediated mitochondrial fission (von Eyss et al., 2015). In other words, there is also a feedback loop between the Hippo/YAP pathway and mitochondrial fission, but the bidirectional effects are indirect, and the mechanism needs to be determined.
Tumor specimen microarrays revealed that the abundance of YAP in the cytoplasm is no less than that in the nucleus (Li et al., 2017). Recent studies also found that YAP located in the cytoplasm and subcellular regions plays a critical role in melanoma and cardiac diseases (Morikawa et al., 2017; Liu et al., 2019a). In human rectal cancer cells, YAP knockout initiates Drp1-related mitochondrial fission and inhibits cell migration by the JNK-Drp1-Htra2/Omi pathway. Coimmunoprecipitation assays found direct interactions between YAP and JNK in the cytoplasm, suggesting that YAP in the cytoplasm may trigger this process (Li et al., 2017). Regrettably, there are few studies about whether YAP in the cytoplasm mediates mitochondrial dynamics in the heart.
Role of the Hippo Pathway in Mitophagy
The mechanism underlying mitophagy—a type of selective autophagy that maintains MQC in the heart—has attracted considerable research attention recently. During mitophagy, impaired mitochondria are selectively fused and removed by lysosomes, and essential components are recycled. Moderate mitophagy plays a cardioprotective role under environmental stress owing to recycling of nutrients, maintenance of energy homeostasis, and elimination of toxic substances, whereas excessive mitophagy results in cardiomyocyte death and cardiac dysfunction (Tahrir et al., 2019). Mitophagy includes Pink1-Parkin-mediated mitophagy, receptor-mediated mitophagy, and mitochondrial dynamics-mediated mitophagy.
Pink1-Parkin-mediated mitophagy is the most canonical form. In intact mitochondria, Pink-1 is continuously degraded in the IMM. However, in depolarized mitochondria, Pink-1 accumulates at the OMM and phosphorylates Mfn2 to recruit and ubiquitinate Parkin, finally binding protein 1 light chain 3 (LC3) through p62 and inducing mitophagy (Saito and Sadoshima, 2015). Several lines of evidence indicate that the Hippo pathway regulates Pink1-Parkin-mediated mitophagy in multiple ways (Li et al., 2018; Yu et al., 2019b; Shang et al., 2020a).
Huang et al. (2011) first discovered in mice and HL-1 cardiomyocytes that Parkin knockout attenuated the cardioprotection of ischemic preconditioning because of decreased mitophagy. This research shows that Parkin-related mitophagy is cardioprotective in MI/RI. However, the molecular mechanisms of Parkin in the cardiac system are still poorly understood. Mst1 has been identified as an upstream mediator of Parkin-mediated mitophagy (Wang et al., 2018b; Shang et al., 2020a). Melatonin, an oxygen radical scavenger involved in cardiac protection, exerts a favorable effect on DCM by enhancing Parkin-mediated mitophagy through inhibition of Mst1 phosphorylation in vivo and in vitro (Wang et al., 2018b).
This conclusion has also been validated in septic cardiomyopathy. Shang et al. revealed that Mst1 is overexpressed in mice with septic cardiomyopathy and that Mst1 deletion can alleviate LPS-induced cardiomyocyte death and improve cardiac function in vitro. At the molecular level, the ablation of Mst1 increases Parkin-mediated mitophagy through the MAPK-ERK signaling pathway in vitro (Shang et al., 2020a). Excessive mitophagy causes cardiomyocyte damage because of the overclearance of mitochondria (Tahrir et al., 2019). In addition, the activation of mitophagy can induce calcium overload, which is harmful to cardiomyocytes (Rusnati et al., 2019). However, the detailed molecular mechanisms need more studies to reach a definitive answer.
Mitochondrial receptor-mediated mitophagy also plays a vital role in the heart. At present, several mitophagy receptors have been identified, including NIX, BNIP3, and FUNDC1 (FUN14 domain-containing protein 1).
Bnip3, an atypical member of the Bcl-2 family of proapoptotic proteins, is mainly located in the OMM to promote mitophagy and apoptosis (O'Sullivan et al., 2015). The Hippo pathway has a close relationship with Bnip3-related mitophagy. Xiang et al. discovered that HIF-1 binds to TAZ directly through the WWTR1 gene, which encodes TAZ, and that these two proteins serve as coactivators for each other. TAZ downregulation markedly reduced the expression of genes targeted by HIF-1, including Bnip3 in human breast cancer cells (Xiang et al., 2015). This was the first study to find an indirect relationship between the Hippo pathway and Bnip3-related mitophagy.
Subsequent studies aimed to explore the mechanism underlying YAP-mediated regulation of Bnip3-related mitophagy. JNK phosphorylation is activated under YAP deficiency, and p-JNK binds the promoter of Bnip3 to increase mitophagy and promote hepatocellular carcinoma metastasis in vitro (Shi et al., 2018). Furthermore, upregulated Mst1 can inhibit Bnip3-related mitophagy through the JNK pathway to inhibit colorectal cancer proliferation and migration in vitro (Li et al., 2018). These studies suggest an intricate reciprocal interplay between mitophagy and the Hippo pathway. In previous studies, Bnip3-related mitophagy has been reported to participate in the development of CVDs (Li et al., 2020; Peng et al., 2020). However, the pathogenic relevance of Bnip3-related mitophagy mediated by the Hippo pathway in CVDs is still unknown.
Zhou et al. (2017) first reported that in response to MI/RI, FUNDC1-mediated mitophagy is activated, which causes mitochondrial fragmentation and cytochrome-C degradation and ultimately inhibits apoptosis signaling in vivo and in vitro. Yu et al. illustrated that in Mst1 KO mice, the cardiac infarction area is reduced in the context of MI/RI. Mechanistically, the loss of Mst1 increases FUNDC1-regulated mitophagy through the MAPK/ERK-CREB pathway in vitro (Yu et al., 2019b). Thus, the Hippo pathway is involved in receptor-related mitophagy. However, more experiments are needed to further assess the relationship between the Hippo pathway and other mitophagy receptors.
Mitochondrial dynamics-related factors act as mediators of mitophagy in cells. Chen and Dorn (2013) showed that Mfn2 is phosphorylated by PINK1 and then promotes the recruitment of Parkin to damaged mitochondria. In the mouse heart, Drp1 deletion inhibits mitophagy, inducing cardiac dysfunction in vivo (Ikeda et al., 2015). Opa1 also activates mitochondrial fusion and mitophagy simultaneously in vitro (Quintana-Cabrera et al., 2018). Therefore, the role of mitochondrial dynamics in regulating mitophagy in cardiac systems seems complex and relevant.
Some of the latest studies have focused on the molecular mechanism by which the Hippo pathway regulates mitochondrial dynamics factor-mediated mitophagy. Yan et al. (2018) illustrated that overexpression of YAP promoted gastric cancer cell migration and survival by mediating the activation of SIRT1-Mfn2-mitophagy signals. The upregulation of Mst1 causes mitochondrial damage under I/R injury in the kidneys, and Mst1 overexpression inhibits Opa1-related mitophagy through the AMPK pathway in vitro (Feng et al., 2018).
In H9C2 cells, the upregulation of LATS2 significantly exacerbated mitochondrial injury and cell death. At the molecular level, LATS2 mediates mitochondrial dysfunction by suppressing mitophagy through inhibition of the Prx3-Mfn2 pathway (Tian et al., 2019). Under stress, overexpression of LATS2 increases mitochondrial dysfunction by producing excessive ROS, inducing mPTP opening, and decreasing ATP production. Mitophagy, a clearance mechanism for dysfunctional mitochondria, has been confirmed to be protective for cardiac cells in heart injury.
Despite recent findings, the in-depth relationship between the Hippo pathway and multiple mitophagy receptors requires additional investigation (Fig. 2).
Role of the Hippo Pathway in Mitochondrial Biogenesis
Mitochondrial biogenesis participates in several CVDs (Picca et al., 2018; Jia et al., 2019). Peroxisome proliferator-activated receptor-α coactivator 1 (PGC-1α), a member of the PGC-1 transcriptional coactivator family, mainly exists in tissues and organs with a high energy demand, including the heart, controlling both mitochondrial biogenesis and energy metabolism (Di et al., 2018). The overexpression of PGC-1α in the adult mouse heart leads to mitochondrial biogenesis disorder-induced DCM. Conversely, knockout of PGC-1α can completely reverse cardiac function (Lehman et al., 2000). In addition, the expression of PGC-1α is activated in rats after exercise training post-MI, increasing mitochondrial biogenesis to decrease cardiomyocyte death (Jia et al., 2019).
The Hippo pathway is able to regulate mitochondrial biogenesis-mediated angiogenesis. Mammoto et al. (2018) showed that YAP1-TEAD signaling controls angiogenesis through PGC-1α in human umbilical vein endothelial cells (HUVECs). Genetic ablation of YAP1 in HUVECs represses the expression of PGC-1α, inhibits mitochondrial biogenesis, and downregulates glycolysis and oxygen consumption. Furthermore, the results of studies on mitochondrial fusion and fission proteins demonstrate that YAP1-TEAD1 signaling enhances oxygen consumption by inducing mitochondrial fusion. Of interest, mitochondrial fission-related proteins are not changed. YAP1-TEAD1 can modulate angiogenesis through PGC-1α-regulated mitochondrial biogenesis and metabolism.
Whether YAP1-TEAD1-PGC-1α signaling mediates mitochondrial dynamics remains to be elucidated. In addition, Parkinson disease protein 2 (Park2) has been identified as a new Hippo-YAP target gene that is also associated with mitochondrial biogenesis (Wang et al., 2018a); however, the detailed mechanism has not been explored (Fig. 2).
The Hippo Pathway Regulates Mitochondrial Apoptosis Under Oxidative Stress
Mitochondrial apoptosis, the intrinsic pathway of apoptosis, is a tightly regulated form of programmed cell death. Moderate mitochondrial apoptosis is required to remove damaged cells and maintain the normal physiological function of the heart. An abnormal level of apoptosis in the heart leads to the pathogenesis of CVDs (Del Re et al., 2019).
ROS, including the superoxide anion, the hydroxyl radical, and hydrogen peroxide, are pivotal signaling molecules that are produced largely by mitochondria and exert diverse effects under both physiological and pathophysiological conditions in the heart (Boengler et al., 2019). Normal levels of ROS regulate cardiac development, maturation, and excitation–contraction coupling under physiological conditions, whereas increased levels of ROS can induce oxidative stress, which eventually causes cardiac cell death through damage to DNA, proteins, lipids, and mitochondria under pathophysiological conditions. Both processes are closely related to different cardiac cell fates, which are determined by the effects of ROS signaling on multiple signaling pathways (Peoples et al., 2019).
In recent years, a growing number of studies have focused on ROS signaling and oxidative stress through mitochondrial apoptosis regulated by the Hippo pathway in a series of CVDs, including MI/RI and DCM (Byun et al., 2019; Francisco et al., 2019).
Phosphorylated Mst1 participates in regulation of the cell cycle and apoptosis by phosphorylating a series of substrates, including LATS1/2, histone H2B, FOXO3, and Bcl-xl (B cell leukemia/lymphoma 2 XL) (Nakamura et al., 2016; Bitra et al., 2017). In addition to LATS1/2, Mst1 can phosphorylate other substrates that trigger the noncanonical Hippo signaling pathway (i.e., not through LATS and YAP) through special signaling complexes formed at multiple subcellular locations. Del Re et al. found that in the case of oxidative stress, Mst1 binds with K-Ras, and Ras association domain family member 1 isoform A RASSF1A (the scaffold of the Ras association domain family A) forms a signaling cassette and colocalizes to mitochondria with activated K-Ras, which induces the activation of Mst1 and increases mitochondrial apoptosis in cardiomyocytes (Fig. 3).
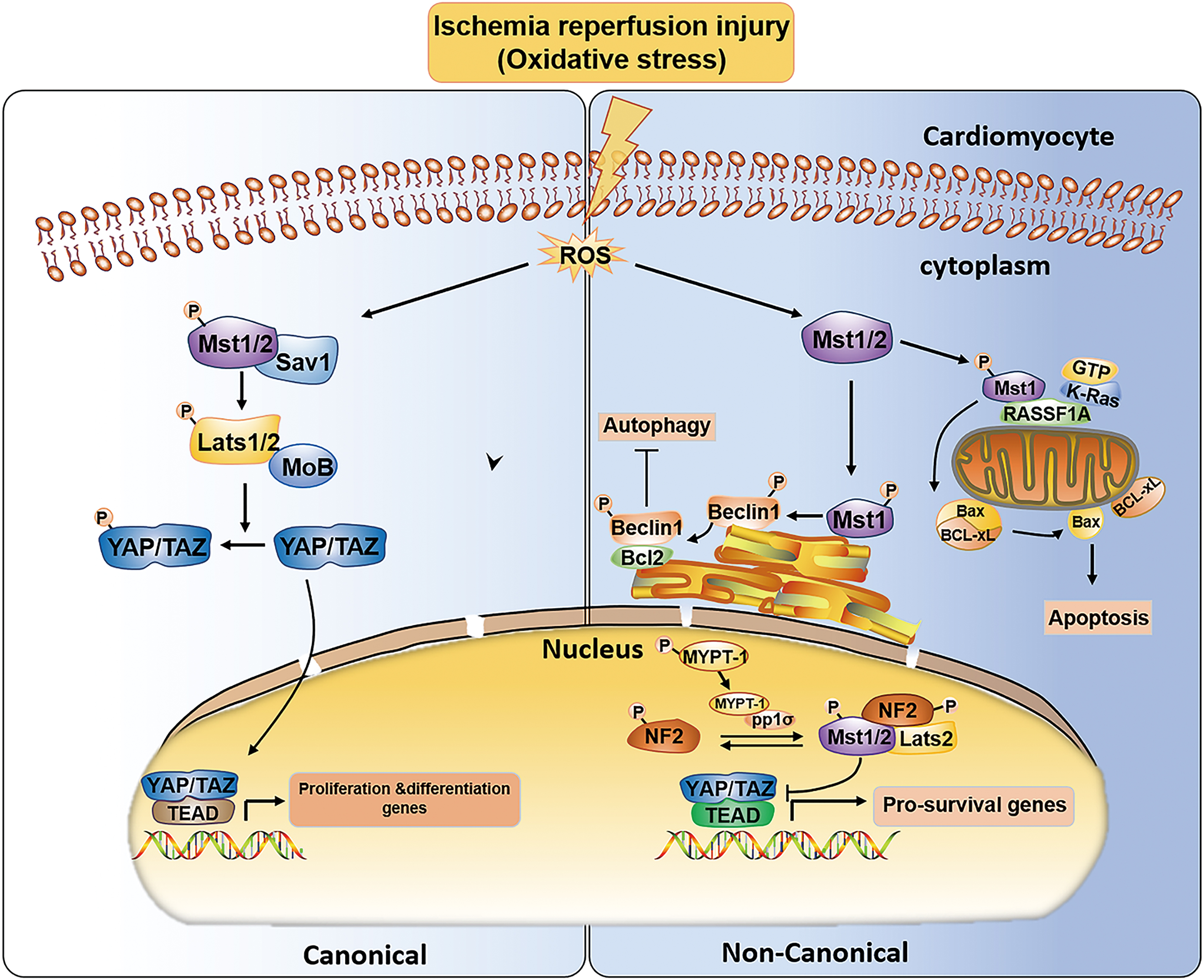
The canonical and noncanonical Hippo pathways under ischemia–reperfusion injury. In the context of oxidative stress, ROS overproduction, on the one hand, activates the canonical Hippo pathway to target proliferation/differentiation genes. Meanwhile, it also activates the noncanonical Hippo pathway to maintain the balance of mitochondrial apoptosis and autophagy. In addition, it initiates the noncanonical Hippo pathway in the nucleus to target prosurvival genes. Bax, B cell leukemia/lymphoma 2-associated protein X; Bcl-xl, B cell leukemia/lymphoma 2 XL; Beclin1, a Bcl-2 homology protein; MYPT-1, myosin phosphatase target subunit-1; NF2, neurofibromin 2; RASSF1A, the scaffold of the Ras association domain family A; ROS, reactive oxygen species. Color images are available online.
RASSF1A acts as a scaffold protein in this signaling cassette and consists of a Ras-associated (RA) domain that interacts with activated K-Ras and a Salvador-Rassf-Hippo (SARAH) domain that binds and phosphorylates Mst1. Then, activated Mst1 directly phosphorylates Ser14 in the BH4 domain of Bcl-xl at the OMM and dissociates Bcl-xl from Bax (B cell leukemia/lymphoma 2-associated protein X) to trigger mitochondrial apoptosis (Del Re et al., 2014). In addition, Nakamura et al. reported that Mst1 is activated by oxidative stress during MI/RI, and cardiac-specific overexpression of Mst1 in transgenic mice (Tg-Mst1) results in congestive heart failure, whereas Mst1 deletion in Tg-DN-Mst1 mice (transgenic mice with cardiac-specific overexpression of dominant-negative Mst1) decreases p-Bcl-xl levels and the infarct area.
In contrast, in knock-in mice (KI) with knockout of phosphorylated Bcl-xl at Ser14, cardiomyocyte mitochondrial apoptosis is reduced and cardiac dysfunction is attenuated. An increase in myocardial mitochondrial apoptosis and fibrosis induced by Mst1 was observed in het KI-Tg-Mst1 mice compared with Tg-Mst1 mice. All these results suggest that the cardioprotective effect of Mst1 against MI/RI is primarily regulated by inhibition of Ser14 phosphorylation of Bcl-xl. Suppression of Ser14 phosphorylation of Bcl-xl reduces mitochondrial apoptosis in cardiomyocytes and improves cardiac function (Nakamura et al., 2016).
Molecular investigations showed that Bax and Bcl-xl are members of the Bcl-2 family; Bcl-xl is an antiapoptotic protein, and Bax is a proapoptotic protein. Under normal conditions, Bax mainly exists in the cytosol and is rarely expressed in the mitochondria. When cells detect an apoptotic stimulus, Bax is translocated to the mitochondria, especially to the OMM; it oligomerizes in the OMM and forms pores to result in mitochondrial outer membrane permeabilization (MOMP), which allows a variety of proteins, such as cytochrome-C, to be released from the mitochondrial intermembrane space into the cytosol and triggers mitochondrial apoptosis. However, the interaction between Bcl-xl and Bax prevents the activation and mitochondrial translocation of Bax (Dai et al., 2016).
Therefore, under oxidative stress conditions, the K-Ras-RASSF1A-Mst1 signaling cassette promotes dissociation of the Bcl-xl-Bax complex and increases mitochondrial apoptosis, which contributes to cardiomyocyte death and deterioration of cardiac function.
In addition, oxidative stress-induced tissue damage is related to the mechanisms of apoptosis and necrosis. The latest studies show that Bak and Bax are associated with mitophagy to remove damaged mitochondria (Bernardini et al., 2019). Furthermore, Bcl-xl can modulate cell death by altering mitochondrial shape and dynamics (Aouacheria et al., 2017). Therefore, all these results illustrate that the proteins of the Bcl-2 family are involved in mitochondria-mediated cell death through various complex mechanisms.
Francisco et al. (2019) found that under oxidative stress conditions, the hearts of mice with a systemic RASSF1A deletion (RASSF1A−/− ) do not have a larger infarction area than those of mice with a cardiomyocyte-specific deletion of RASSF1A (RASSF1A KO), which may be linked to inflammation. Inflammatory factors such as TNF-α are significantly increased in RASSF1A KO mice compared with RASSF1A−/− mice. These data indicated that these inflammatory factors may be secreted by nonmyocytes. In macrophages, the overexpression of RASSF1A markedly reduced inflammatory factor expression by inhibiting YAP phosphorylation. In other words, RASSF1A/Mst1 signaling can activate both the canonical Hippo pathway to reduce inflammatory reactions and the noncanonical Hippo pathway to promote mitochondrial apoptosis.
Therefore, RASSF1A/Mst1 signaling may play an important role in the heart through different pathological factors in a cell type-dependent manner in the presence of oxidative stress, and selective inhibition of RASSF1A/Mst1 in myocardial cells may be the most appropriate treatment strategy.
The Hippo Pathway Regulates Lipid Metabolism in Mitochondria
The dysregulation of lipid metabolism, especially lipoxidation, has been recognized as a hallmark of CVDs (Gianazza et al., 2019). The association of the Hippo pathway with lipid metabolism has recently been described (Koo and Guan, 2018; Tharp et al., 2018; Liu et al., 2019b). YAP participates in lipid accumulation and steatosis in the liver (Koo and Guan, 2018). In hepatocellular carcinoma cell lines, YAP activation increases lipogenesis (Liu et al., 2019b). The Hippo pathway also acts on the final phase of lipid metabolism in mitochondria. Tharp et al. (2018) showed that YAP can directly promote the transcription of uncoupling protein 1 (UCP1) to accelerate the breakdown of brown adipocytes, increase heat production, and reduce ATP biosynthesis (Fig. 4). This result suggests that the role of the Hippo pathway in adipose tissue metabolism is fat cell-type dependent.
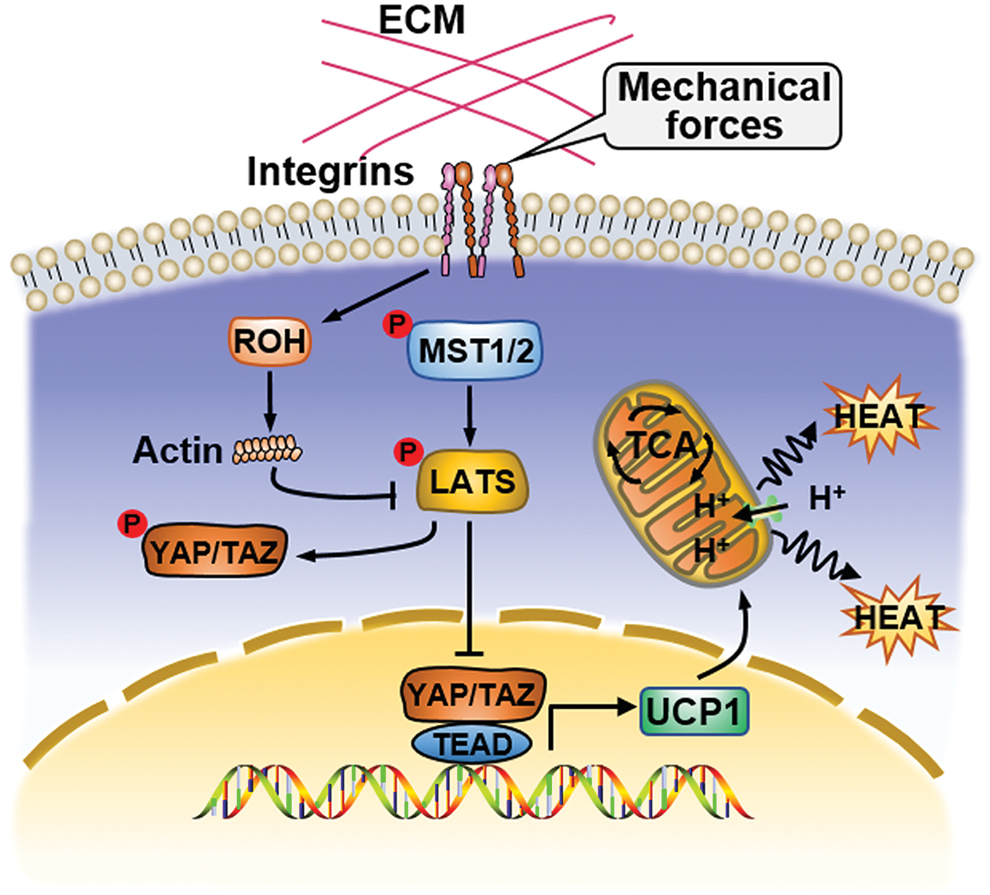
The Hippo pathway regulates lipid metabolism in mitochondria. Inactivation of the Hippo pathway can promote nuclear translocation of YAP, and YAP then combines with TAZ and TEAD to promote the transcription of UCP1 to accelerate the breakdown of brown adipocytes, increase heat production, and reduce ATP biosynthesis. TCA cycle, tricarboxylic acid cycle; UCP1, uncoupling protein 1. Color images are available online.
In addition, mitochondria are a key organelle for catabolic and anabolic processes of glucose and glutamine, which can be targeted by the Hippo pathway (Koo and Guan, 2018). Whether the Hippo pathway participates in the metabolic processes of glucose and glutamine in mitochondria requires further investigation.
The Hippo Pathway Orchestrates MQC in MI/RI
Cardiac I/R injury caused by the blockage of coronary vessels induces acute tissue hypoxia and cellular energy deprivation and activates oxidative stress in the heart, leading to irreversible cardiomyocyte death and decreased heart function. At present, treatments for MI/RI mainly include ischemic conditioning and primary percutaneous coronary intervention (Heusch, 2018). During the early stages of myocardial ischemia, cardiac hypoxia restricts OXPHOS, decreases ATP production, increases mitochondrial calcium influx, and activates ROS-mediated signaling, all of which are responsible for MPTP-induced mitochondrial depolarization and swelling, and ultimately cell death. Thus, there are tightly regulated mechanisms by which MQC mitigates I/R heart pathology.
In a previous study, the Hippo pathway was involved in myocardial repair and regeneration after MI/RI (Heallen et al., 2013). Through diverse upstream regulators, such as neurofibromin 2 (NF2) and RASSF1A, various components of the Hippo pathway are activated in a canonical or noncanonical manner and mediate different functional responses to cardiac I/R injury (Mia et al., 2016; Nakamura et al., 2016). MI/RI-induced stress triggers the phosphorylation cascade of Mst1 and LATS2 in the cytoplasm and promotes the nuclear translocation of the downstream effectors YAP and TAZ, ultimately regulating myocardial proliferation and differentiation, which is the canonical Hippo pathway.
On the contrary, activated Mst1 can phosphorylate substrates other than canonical Hippo pathway components or activate the downstream Hippo pathway in abnormal subcellular locations, eventually regulating prosurvival genes or apoptosis and autophagy proteins through a noncanonical Hippo pathway (Matsuda et al., 2016; Mia et al., 2016; Nakamura et al., 2016) (Fig. 3).
There has been accumulating interest in the Hippo pathway and its potential association with MQC in the context of MI/RI (Wang and Song, 2018; Ma and Dong, 2019; Yu et al., 2019b). During the cardiac I/R injury period, the balance of mitochondrial dynamics is disturbed. Suppression of mitochondrial fragmentation and a moderate increase in mitochondrial fusion may reverse the pathological progression of MI/RI. Downregulation of Mst1 attenuates renal I/R injury in gene-modified mice by inhibiting mitochondrial fission (Li et al., 2019). Similarly, knockout of Mst1 also inhibits mitochondrial fission and reduces cardiomyocyte apoptosis, eventually leading to remission of cardiac dysfunction after I/R (Wang and Song, 2018). There are also studies demonstrating that overexpression of YAP improves heart function by upregulating OPA1-related mitochondrial fusion during MI/RI (Ma and Dong, 2019).
In some diseases, the Hippo pathway downstream effector YAP influences mitochondrial fission, but this correlation was not observed in MI/RI (Li et al., 2017; Huang et al., 2018). However, in a recent study, cardioprotection was found only when Drp1 was suppressed in the early phase of reperfusion; interestingly, cell death was aggravated when mitochondrial fission was targeted for inhibition during reoxygenation (Dong et al., 2016). This illustrates the complicated mechanisms of mitochondrial dynamics and their impact on cardiac I/R injury.
During ischemia, upregulation of mitophagy has been proven to provide protection. This conclusion was also confirmed in a recent study, and the increase in mitophagy could alleviate cardiomyocyte injury in Mst1 KO mice suffering from MI/RI (Wang and Song, 2018). However, some studies showed that the reduction of mitophagy may have cardioprotective effects under I/R, probably because of inhibition of excessive degradation of malfunctioning mitochondria (Ji et al., 2016). The specific mechanism underlying the different functions of mitophagy in various stages of MI/RI requires further research.
In the case of I/R, apoptosis can cause myocardial death and decrease cardiac performance (Matsuda et al., 2016). Mitochondrial apoptosis, the intrinsic pathway of apoptosis, occurs in advance of the extrinsic pathway (Li and Liu, 2018). Mst1 acts as an important regulator of mitochondrial apoptosis in cardiac I/R injury. Activated Mst1 translocates into the OMM and dissociates the Bcl-xl-Bax complex, thereby inducing mitochondrial apoptosis through the noncanonical Hippo pathway (Nakamura et al., 2016). We speculate that mitochondrial apoptosis can be regulated by YAP because YAP directly mediates OPA1-related mitochondrial fusion and mitochondrial dynamics through the upstream regulatory factors of mitochondrial apoptosis (Ma and Dong, 2019). In summary, the Hippo pathway may be a potential target for I/R treatment in the heart.
The Hippo Pathway Orchestrates MQC in Septic Cardiomyopathy and DCM
Mitochondrial dysfunction is a potentially pathological mechanism that induces septic cardiomyopathy and DCM. The primary mechanisms that participate in abnormal mitochondrial morphology and function in septic cardiomyopathy include excessive ROS production, calcium overload, and altered cAMP-PKA signaling. The insufficiency of mitophagy and abnormal mitochondrial dynamics caused by ROS may be crucial for cardiac systolic function and mitochondrial malfunction in the heart with septic cardiomyopathy (Durand et al., 2017).
YAP overexpression reversed cardiomyocyte damage and was linked to reduced mitochondrial fission (Yu et al., 2019a). Mst1 has also been found to be an upstream regulator of Drp1-related mitochondrial fission and cell apoptosis triggered by F-actin in septic cardiomyopathy (Shang et al., 2020a, 2020b). Downregulation of Mst1 induces cardioprotection in the septic heart by increasing mitophagy (Shang et al., 2020a). These studies suggested that the Hippo pathway may play a key role in septic cardiomyopathy by maintaining mitochondrial homeostasis. The possible mechanisms that may be involved are restoration of the mitochondrial membrane potential, decreased generation of mitochondrial ROS, and repression of mitochondrial apoptosis.
Several mechanisms have been implicated in mitochondrial dysfunction in DCM, including the uncoupling of mitochondria induced by fatty acids, imbalance of mitochondrial dynamics, increased ROS production, impaired mitochondrial calcium homeostasis, and mitochondrial renewal (Gollmer et al., 2020). In DCM mice, diabetes compromised heart mitochondrial function, which was alleviated in Mst1 knockout mice by enhancing mitophagy to remove damaged mitochondria in cardiomyocytes (Zhang et al., 2017b; Wang et al., 2019). The number of mitochondria was also reduced by knocking out Mst1 in diabetic mice, and the expression of PGC-1α was increased (Wang et al., 2019). Accordingly, we hypothesize that the number of mitochondria is partly regulated by Mst1-mediated mitochondrial biogenesis.
Conversely, the nuclear translocation of YAP increases in the diabetic heart, which suppresses cardiac fibrosis and induces the dedifferentiation of myocardial cells (Liu et al., 2020). In many heart diseases, YAP functions in numerous pathological processes by regulating MQC (Ma and Dong, 2019; Yu et al., 2019b). Whether YAP contributes to the development of DCM by mediating mitochondrial morphology and function requires further investigation.
Conclusions
In recent years, extensive effort has focused on research on the contribution of MQC to CVDs. Mitochondria are important cellular organelles in the heart because they supply energy, serve as the main producer of ROS and target of oxidative stress, and control the final phase of major nutrient metabolism. An accumulating number of studies have indicated that Hippo pathway activation could influence various aspects of MQC to regulate the role of cardiomyocyte apoptosis, proliferation, and antioxidative activity, which may play a critical role in the pathology of CVDs. Therefore, intervening in the interaction between the Hippo pathway and MQC may be a promising novel strategy for CVD.
The hearts of mammals, including humans, are known to have a poor regenerative capacity, and most patients with CVDs will advance to heart failure in later stages, ultimately leading to death. Inactivation of the Hippo pathway in the myocardium of adult mice improves cell survival and cardiac function after MI (Meliambro and Campbell, 2018). Further studies found that Hippo pathway activation could inhibit mitophagy and promote mitochondrial fission, both induced by oxidative stress, to attenuate cardiac dysfunction after MI/RI (Zhang and Yu, 2018; Yu et al., 2019b). Similarly, the Hippo pathway is turned off, resulting in an increase in cardiomyocyte contractility and a reduction in cardiac injury in mice with DCM by coordinating mitophagy and apoptosis (Yang et al., 2018). In the myocardium of mice with septic cardiomyopathy, inhibiting the Hippo pathway induces cardioprotection by regulating MQC under inflammatory stress (Yu et al., 2019a). At present, a number of drugs are available to target the Hippo signaling pathway in various types of cancer, and this pathway may be an attractive target for CVDs.
However, the specific mechanisms underlying the cardioprotection regulated by the interaction between the Hippo pathway and MQC have not yet been identified. For example, knockout of Mst1 favors cardiomyocyte survival but is simultaneously harmful to the heart because it promotes fibroblast proliferation. Further investigation is needed to understand the separate roles of the Hippo pathway in nonmyocytes in the heart. In addition, the Hippo pathway contributes to cardioprotection by both promoting cardiomyocyte proliferation and coordinating mitophagy and apoptosis. The challenge for future investigations is that whether the regulation of those biological effects occurs through a canonical or noncanonical pathway or complex signaling remains unclear.
Footnotes
Disclosure Statement
There are no conflicts of interest.
Funding Information
This work was supported by the grants from the National Natural Science Foundation of China (81770490).