
Abstract
Head and neck squamous cell carcinoma (HNSCC) is a malignancy with relatively high incidence and poor prognosis. RNA-binding proteins (RBPs) were reported to be dysregulated in multiple cancers and were closely associated with tumor initiation and progression. However, an integrated analysis of the roles of RBPs in HNSCC has not been conducted. In the present study, we obtained transcriptome data and corresponding clinical information of HNSCC patients from The Cancer Genome Atlas database and screened out differentially expressed RBPs between tumor and normal tissues. Subsequently, we utilized a series of bioinformatics analyses to elucidate the potential functions and prognostic value of these RBPs in HNSCC. As a result, a total of 88 aberrantly expressed RBPs were identified, including 63 downregulated and 25 upregulated RBPs. Functional enrichment analysis suggested that the differentially expressed RBPs mainly participated in mRNA metabolic processes, RNA processing, RNA transport, regulation of RNA stability, RNA degradation, and mRNA surveillance pathway. Three RBP genes (NOVA1, EZH2, and RBM24) were determined as prognosis-related hub genes from which EZH2 and NOVA1 were selected to construct a prognostic signature based on LASSO Cox regression algorithm. Further analysis demonstrated that the high-risk patient group stratified by the risk signature has advanced tumor grade and poorer overall survival when compared with low-risk group. Moreover, univariate analysis showed that the risk score, tumor stage, T stage, and N stage were significantly associated with patient overall survival and the multivariate analysis results indicated that the risk score and age were greatly correlated with patient prognosis. Overall, this study provided a comprehensive landscape of RBPs in HNSCC and identified an effective gene signature for predicting the clinical outcomes of HNSCC patient, which may contribute to clinical decision making and individualized cancer treatment.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is ranked as the sixth most common cancer worldwide with the 5-year survival rate remaining around 50% (Scott et al., 2005). In spite of great advances in the development of innovative treatments over the past few decades, there has not been effective therapies for HNSCC patients. Therefore, a comprehensive analysis of significant genes during HNSCC tumorigenesis is greatly needed, which may help to identify promising diagnostic biomarkers and therapeutic targets for HNSCC.
RNA-binding proteins (RBPs) are a group of proteins containing RNA-binding domains and have been intimately involved in gene expression regulation (Burd and Dreyfuss, 1994). It mainly consists of cotranscriptional and posttranscriptional gene regulation, including RNA maturation, RNA turnover, RNA localization, and translation (Glisovic et al., 2008). A published study has reported 1542 kinds of RBPs through high-throughput screening in human cells (Gerstberger et al., 2014). These RBPs were demonstrated to mediate cell physiology and a variety of biological processes, such as RNA splicing, mRNA stability, localization, and protein translation. Furthermore, it is increasingly recognized that dysregulation of RBPs is closely associated with the initiation and progression of various human cancers.
More importantly, numerous studies have reported the importance of RBPs in HNSCC tumorigenesis. For example, a study demonstrated that RBP FXR1 could mediate cellular senescence by mRNA turnover pathway. Specifically, FXR1 could bind and destabilize p21 mRNA so as to reduce p21 protein expression in oral cancer cells and the underlying mechanism through regulating p53-dependent manners (Majumder et al., 2016). Another study found the positive correlation between high hnRNP K expression and clinicopathological parameters, which represent bad treatment outcomes. Multivariate analyses suggested that hnRNP K had the potential of predicting the overall survival rate of oral squamous cell carcinoma patients independently, acting as promising prognostic and therapeutic markers (Wu et al., 2012). RBP HuR participates in the regulation of various kinds of cellular functions such as tumor cell proliferation, apoptosis, invasion, and metastasis. Under hypoxia environment, it was indicated that HuR knockdown led to elevated c-Myc protein expression. These findings revealed a HuR-mediated posttranscriptional mechanism, which controlled c-Myc expression in oral cancer progression (Talwar et al., 2011). What is more, overexpression of Lin28b was observed in HNSCC and it was demonstrated to remarkably promote tumor progression through modulating multiple gene targets, implying the biological importance of RBPs in HNSCC development and progression (Alajez et al., 2012).
Taken together, these studies focused on one specific RBP and analyzed its roles in HNSCC occurrence and progression. However, a systematic analysis of RBPs may present novel and comprehensive insights into the underlying mechanisms during cancer progression. The development of next-generation sequencing techniques serves as a valuable mean to provide new insights into the abnormal gene alteration during tumor initiation and progression (Kamps et al., 2017). Moreover, the application of these high-throughput methods facilitates the identification of promising biomarkers for cancer diagnosis and prognosis evaluation (Yohe and Thyagarajan, 2017). The Cancer Genome Atlas (TCGA) database is a public platform which collects a large number of transcriptome data (Tomczak et al., 2015). Bioinformatics analyses based on high-throughput data from public database could get a relatively reliable and precise results by large clinical sample size.
In this study, we downloaded available data from TCGA database and conducted a series of bioinformatics analysis to detect the potential molecular functions and clinical significance of RBPs in HNSCC. We finally identified a multiple of differentially expressed RBPs underlying HNSCC pathogenesis and constructed an effective gene signature to predict patient prognosis. Our findings may provide innovative mechanism underlying HNSCC carcinogenesis and lay a foundation for developing novel strategies for HNSCC treatment and prognosis evaluation.
Materials and Methods
Differentially expressed RBPs identification
Transcriptome data of HNSCC and compared normal samples with corresponding clinical information were obtained from TCGA GDC portal. The sample ID of each clinical sample was listed in the Supplementary Data. We chose the FPKM (The Fragments per Kilobase of transcript per Million mapped reads) data, which means the data have been normalized by TCGA database through dividing it by the gene length and the total number of reads mapped to protein-coding genes. We used hg38 to annotate the genes and no data were excluded from the analysis. Detailed clinical characteristics of HNSCC patients are summarized in Table 1. Differential expression analysis was conducted on profiles of 1542 RBPs using the Limma package in R software (version 3.6.1). The choice of 1542 RBPs was based on high-throughput screening analysis in human cells by a published study (Gerstberger et al., 2014). Genes that conformed to the threshold of adj. p-Value <0.05 and |logFC| > 1.0 were defined as differentially expressed RBPs.
Baseline Patient and Primary Tumor Characteristics (n = 515)
Gene ontology and Kyoto Encyclopedia of Genes and Genomes functional enrichment analysis
Gene ontology (GO) enrichment, which consists of three main terms: biological process, cellular component, and molecular function, was performed to explore the biological functions of these differently expressed RBPs. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was utilized to detect potential enriched pathways of differentially expressed RBPs. Functional enrichment analysis was conducted by the clusterProfiler package in R (version 3.6.1).
Protein-protein interaction network construction and module screening
The differently expressed RBPs were mapped to the STRING database to create an interactive network, presenting associations among selected genes. The Cytoscape software was then used to visualize protein-protein interaction network. The significant gene modules were selected by Molecular Complex Detection (MCODE) plugin and cytoHubba plugin was used to identify hub gene in this comprehensive network.
Prognostic value of RBPs
The differentially expressed RBPs were subjected to survival analysis to detect their potential prognostic value. Prognostic-related RBPs then underwent LASSO Cox regression analysis and a risk signature was constructed based on the results. We utilized glmnet package in R software to perform LASSO regression analysis. The risk score of individual sample was calculated by the established risk model and we identified the median risk score of all the HNSCC patients. Then, patients with a risk score larger than median were assigned to high-risk group, and patients with a risk score lower than the median value were stratified into low-risk group. Kaplan–Meier survival curves were plotted to compare the overall survival of two different patient groups. We used the survival package in R to perform the survival analysis. Additionally, univariate and multivariate Cox regression analyses were performed to determine the potential of the risk signature acting as an independent prognostic indicator. p < 0.05 was defined as statistically significant through all the analyses.
Results
Identification of differentially expressed RBPs
Transcriptome data and clinical features of 515 HNSCC samples and 43 compared normal samples were downloaded from TCGA. Detailed clinical information of HNSCC samples is concluded in Table 1. This study focused on a list of 1542 RBPs and 88 differentially expressed RBPs were finally screened out (25 upregulated and 63 downregulated RBPs). All the detailed information of differentially expressed RBPs, including fold change and adj. p-Value is summarized in Supplementary Table S1. Volcano plot showing the distribution of upregulated and downregulated RBPs is plotted in Figure 1A. A heatmap presenting the expression profiles of differentially expressed RBPs in HNSCC and compared normal samples is shown in Figure 1B.
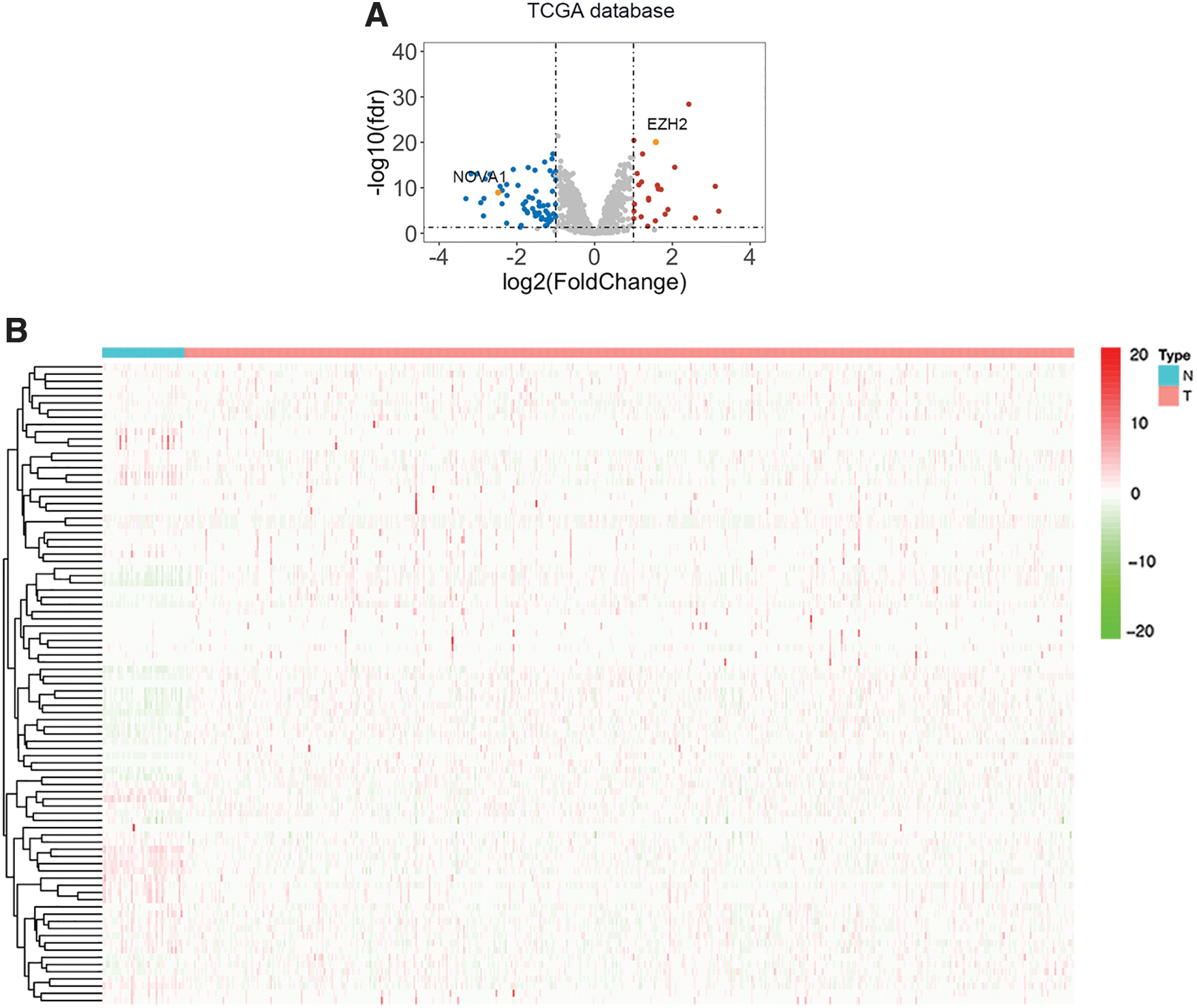
Differential analysis of 1542 RBPs in HNSCC.
Functional enrichment analysis of differentially expressed RBPs
Differentially expressed RBPs were subjected to functional enrichment analysis. GO analysis results indicated that differentially expressed RBPs were primarily enriched in biological functions, including translation regulation, mRNA processing, regulation of mRNA metabolic process, and regulation of RNA stability (Fig. 2A). The enriched terms of cellular component showed that differently expressed RBPs were closely associated with ribonucleoprotein granule, cytoplasmic stress granule, and RNA cap-binding complex (Fig. 2A). For molecular function, the selected RBPs were mainly enriched in mRNA binding, translation regulator activity, and mRNA regulatory element binding (Fig. 2A). Meanwhile, results of KEGG pathway analysis suggested that differentially expressed RBPs were involved in RNA transport, microRNAs in cancer, mRNA surveillance pathway, and RNA degradation pathways (Fig. 2B).

Functional enrichment of differentially expressed RBPs.
PPI network construction and significant gene modules
To have a comprehensive insight into the differentially expressed RBPs, we constructed an interactive PPI network using Cytoscape software (Fig. 3). As was shown, orange indicates upregulated RBPs and blue indicates downregulated ones. MCODE tool was used to figure out significant gene modules and cytoHubba plugin was utilized to identify hub gene. As is shown in Figure 4A–C, 3 significant gene modules were screened out and 10 hub gene with the most connective degree was identified (Fig. 4D). Specifically, module 1 consists of 6 nodes and 13 edges, module 2 was made up of 10 nodes and 22 edges, whereas 4 nodes and 5 edges constitute module 3. Regarding hub genes, the most significant gene was ELAVL4 with the score of 12, followed by ELAVL3 (score = 11), NOVA1, and PIWIL1 (score = 10).

PPI network construction and hub gene identification. A comprehensive PPI network of differentially expressed RBPs was constructed. Orange indicates upregulated RBPs and blue indicates downregulated ones. PPI, protein-protein interaction. Color images are available online.

Module analysis and hub gene identification.
Prognostic value of hub RBPs and risk signature construction
To evaluate the prognostic value of hub RBPs on HNSCC, we performed survival analysis and found out three prognostic-related hub RBP genes (NOVA1, EZH2, and RBM24) (Fig. 5). Afterward, LASSO Cox regression algorithm was conducted and NOVA1 and EZH2 were selected to construct a predictive model, which is indicated in Figure 1A. Then, a survival analysis was applied to assess the predictive efficacy. HNSCC patients were divided into low- or high-risk subgroups according to the median risk score. Survival analysis results showed significant differences regarding the survival rate between two different patient groups (Fig. 6A), implying the independent predictive value of the risk model on patient clinical outcomes. Moreover, patients in the high-risk group had more advanced tumor grade (Fig. 6B), implying the role of NOVA1 and EZH2 in HNSCC progression.

Kaplan–Meier survival analysis of the prognostic value of NOVA1, EZH2, and RBM24 in HNSCC. Color images are available online.
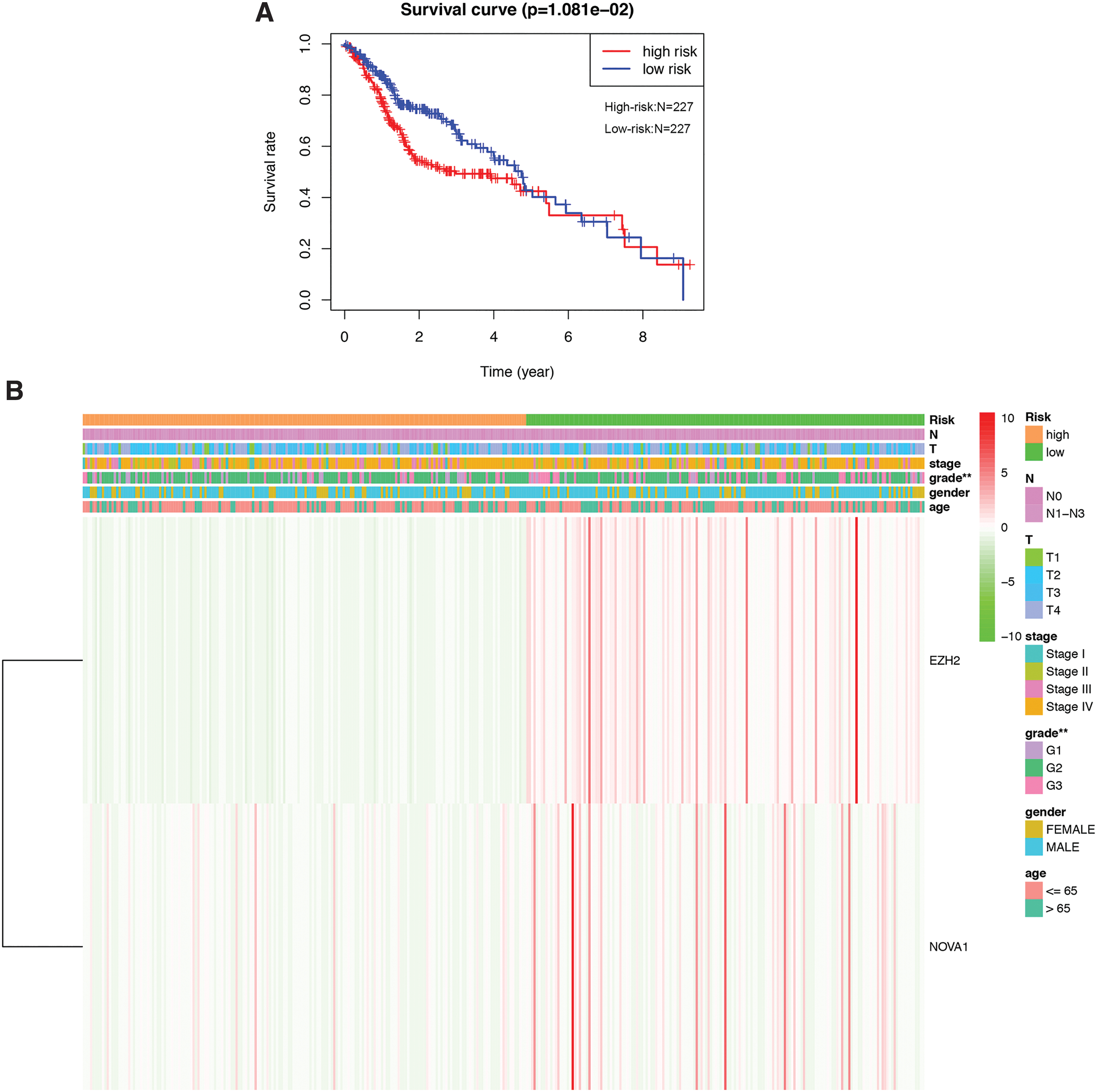
Risk signature constructed for predicting HNSCC prognosis.
Finally, univariate and multivariate Cox regression analyses were conducted to detect potential independent prognostic indicators. The results of the univariate analysis suggested that the risk score, tumor stage, T stage, and N stage were significantly associated with patient overall survival (Fig. 7A). As the risk score, tumor stage, T stage, and N stage increases, the death risk increases. The multivariate Cox regression analysis results indicated that the risk score and age were greatly correlated with patient prognosis (Fig. 6C). Overall, the risk signature we constructed could be utilized as a promising prognostic indicator for HNSCC patients.

Univariate analysis and multivariate analysis of potential biomarkers for HNSCC patients.
Discussion
During the past few decades, great efforts have been put into the development of advanced diagnostic and treatment methods. However, the overall mortality rates of HNSCC cases primarily remained unchanged. As is known, malignant tumors are characterized by uncontrolled cell growth, which mainly results from the dysregulation of driver genes that regulate cell biological processes. Therefore, exploring significant genes and understanding the potential molecular mechanism underlying HNSCC carcinogenesis is necessary and of great value. In recent years, a multiple of studies have suggested the crucial roles of RBPs in the development and progression of various human cancers (Kudinov et al., 2017; Pereira et al., 2017; Dong et al., 2019). However, a comprehensive analysis of the potential functions of RBPs on HNSCC tumorigenesis has not been performed.
In the present study, we provided a well-rounded landscape of 1542 RBPs in HNSCC and illustrated potential molecular mechanisms underlying tumorigenesis and promising risk signature for prognosis evaluation. The functional enrichment analysis of the differentially expressed RBPs showed that RBPs were significantly enriched in translation regulation, mRNA processing, regulation of mRNA metabolic process, and regulation of RNA stability, which confirmed the significant roles of RBPs as posttranscriptional regulators. A multiple of studies during the past decade have verified the potential of mRNA as a promising therapeutic target for malignant diseases. To be more specific, mRNA alterations could lead to the initiation and progression of different cancers and dysregulation of RNA processing and translation may account for cancer pathogenesis (Abdel-Wahab and Gebauer, 2018). Therefore, targeting mRNA processing has been recognized as a potential anticancer strategy (Desterro et al., 2020). For example, transcription factor MYC, which not only modulates RNA processing, but also influences RNA metabolism, is suggested to be an oncogene whose expression is dysregulated in human cancers. Regulating RNA processing through targeting MYC is regarded as an innovative method for treating cancers (Koh et al., 2016). Moreover, RNA stability is crucial for cell life, including cell death and aging, so RBPs involved in regulating RNA stability may affect cancer progression as well (Falcone and Mazzoni, 2018).
Aberrant regulation of RBP-mediated stability programs is frequently observed in multiple cancers. A study found that RBPs mediate renal cancer transcriptome through modulating mRNA stability and regulating cancer-related pathways (Perron et al., 2018). Furthermore, results of KEGG pathway analysis suggested that differentially expressed RBPs participate in RNA transport, miRNAs in cancer, mRNA surveillance pathway, and RNA degradation pathways. Specifically, alterations of crucial pathways underlying mRNA nuclear export could result in genome instability and cancer initiation, serving as a novel therapeutic strategy (Hautbergue, 2017). MiRNAs are a class of noncoding RNA molecules that play essential roles in various aspects of biological phenotypes, including cell proliferation, apoptosis, invasion, and angiogenesis (Bartel, 2004). Functional studies have confirmed that miRNA dysregulation is one of the most common causes for cancer cases (Rupaimoole and Slack, 2017). Due to the fact that miRNAs act as crucial players in the etiology and progression of malignant tumors, RBPs regulating miRNAs in cancer may be exploited as a tool for cancer therapy (Lee and Dutta, 2009). What is more, RNA degradation is considered as a critical posttranscriptional regulatory checkpoint that maintains cellular integrity and homeostasis. Given the close associations between RNA degradation and human cancers, RBPs involved in modulating RNA degradation may function as key actors in carcinogenesis (Saramago et al., 2019).
Subsequently, we identified three prognostic-related hub RBP genes (NOVA1, EZH2, and RBM24) and constructed an effective prognosis signature. A multiple of published research have identified promising risk signature for predicting cancer prognosis. For example, a study presented a nine-RBP gene signature (A1CF, EIF2B5, LSM1, LSM7, MBNL2, RSRC1, TRMU, TTF2, and ZCCHC5), which showed good predictive value for the prognosis of lung squamous cell carcinoma and gained novel insights into the tumor initiation and progression (Li et al., 2020c). Another study implemented a systematic exploration of the expression and prognostic value of RBPs by bioinformatics analyses and established an eight RBPs coding gene-based risk model for lung adenocarcinoma, which may contribute to the development of new treatment targets and prognostic molecular markers (Li et al., 2020a). In glioma, a series of analyses were performed to provide a valuable RBP resource during tumor progression, which revealed promising molecular candidates that were intimately involved in glioma tumorigenesis and had the potential of serving as cancer therapy targets (Wang et al., 2019). What is more, potential markers were screened out from differently expressed RBPs to predict cancer progression in lung cancer, and ROC curve analysis further confirmed that these selected hub genes had relatively good diagnostic efficacy to distinguish lung adenocarcinoma (Li et al., 2020b).
Collectively, our study provides innovative insights into the roles of RBPs during HNSCC development and progression and established a promising prognostic model to evaluate patient prognosis. Nevertheless, this study had several limitations which could not be ignored. First, our results were based on the bioinformatics analysis of the TCGA data and were not validated in clinical patient cohort or other public databases. Second, our study was a retrospective research and a prospective study needs to be implemented to verify the findings. Finally, some lack of clinical parameters in the datasets may decrease the statistical validity and reliability.
In summary, we performed a series of bioinformatics analyses to systemically investigate the functions and prognostic value of differently expressed RBPs on HNSCC. A prognostic model composed of two RBP coding genes was constructed to serve as an independent prognostic indicator for HNSCC. To our knowledge, this is the first report of developing a RBP-associated prognostic model for HNSCC. Our findings may greatly contribute to the understanding of HNSCC pathogenesis and the development of promising treatment targets and prognostic biomarkers.
Footnotes
Authors' Contributions
J.Y. and Q.X. designed the study protocol. J.Y. analyzed the data and Q.X. performed statistical analysis. J.Y. and Q.X. completed the figures. J.Y. wrote the article and Q.X. made revisions. All authors approved the final version of the article.
Disclosure Statement
No competing financial interest exists.
Funding Information
This work was supported by the National Key R&D Program of China (Grant Nos. 2017YFC 0840100, 2017YFC 0840110).
Supplementary Material
Supplementary Data
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.