
Abstract
The Dof transcription factor is a plant-specific transcriptional regulator that plays important roles in plant development and acts as a mediator in plant external stress responses. However, Dofs have previously been identified in several plants but not in alfalfa (Medicago sativa L.), one of the most widely cultivated forage legumes. In the present study, a total of 40 MsDof genes were identified, and the phylogenetic reconstruction, classification, conserved motifs, and expression patterns under abscisic acid (ABA), cold, heat, drought and salt stresses of these Dof genes were comprehensively analyzed. The Dof genes family in alfalfa could be classified into eight classes. Gene ontology (GO) and tissue-specific analysis indicated that most MsDof genes may be involved in biological functions during plant growth. Moreover, the expression profiles and quantitative real-time PCR analysis indicated that eight candidate abiotic tolerance genes were induced in response to four abiotic stresses. This study identified the possibility of abiotic tolerance candidate genes playing various roles in stress resistance at the whole genome level, which would provide new information on the Dof family in alfalfa.
Introduction
Transcription Factors (TFs) are a group of proteins that control cellular processes by binding to specific promoter regions (cis-acting elements) to regulate the expression of downstream target genes (Qu and Zhu, 2006). TFs play fundamental roles in almost all biological processes, such as growth, development and the response to environmental stresses (Jin et al., 2014; Riaño-Pachón et al., 2007). The Dof (DNA binding with one finger) family is a major class of plant-specific TFs that was first found in maize (Yanagisawa, 2002; Yanagisawa and Izui, 1993). Dof genes appear to be more recent in origin than other TFs in plants; the number of Dof members is lower than those of most other TF classes in the plant genome and shows little diversity among different plant species (Shu et al., 2015). For example, there are 36 Dof members in Arabidopsis, 30 in rice (Lijavetzky et al., 2003), 28 in sorghum (Kushwaha et al., 2011), 42 in Medicago truncatula (Shu et al., 2015) and 78 in soybean (Guo and Qiu, 2016). The Dof proteins are characterized by a highly conserved DNA-binding motif containing a single Cys2/Cys2 (C2/C2) zinc finger that consists of an N-terminal Dof domain of 50-52 amino-acid residues; the Cys2/Cys2 Zn2+ finger structure specifically recognizes the 5′-(T/A)AAAG-3′ sequence in the promoter region of target genes as the recognition core (Yanagisawa, 2002; Guo and Qiu, 2016). There is a transcriptional regulation domain at the C-terminal of the Dof proteins that functions in interactions with various regulatory proteins and the activation of gene expression (Ma et al., 2015). As described in a previous study, both the N- and C-terminal regions of Dof proteins may interact with various regulatory proteins or intercept signals to mediate the activation or repression of target genes (Noguero et al., 2013). Moreover, the Dof domain is a bi-functional protein domain that mediates not only DNA binding but also protein-protein interactions (Yanagisawa, 2004), suggesting functional diversity in distinct plant species.
After the Dof domain protein was isolated from maize, the Dof TFs were reported to play important roles in many plant biological processes, such as stress responses, seed germination, flower induction, plant nitrogen assimilation, photosynthesis and the accumulation of seed storage proteins (Venkatesh and Park, 2015; De Paolis et al., 1996). In Arabidopsis, the expression levels of three Dof genes (OBP1, OBP2, and OBP3) are up-regulated under auxin, salicylic acid and cycloheximide treatment (Skirycz et al., 2006; Skirycz et al., 2008; Kang and Singh, 2000); the OBP proteins exhibit similar in vitro DNA-binding properties and are able to interact with OBF4, a bZIP transcription factor (Kang and Singh, 2000). In tea plants, Hui Li and colleagues showed that most Dof genes respond to heat, cold, salt and drought stress treatments (Li et al., 2016); in addition, two Dof genes, TaDof14 and TaDof15, are significantly up-regulated in response to drought treatment (Shaw et al., 2009). Five tomato genes, SlCDF1–5, which are homologous to Arabidopsis Cycling DOF Factors (CDFs), are induced in response to cold, salt, osmotic and heat stresses (Corrales et al., 2014). In potato, most StDof genes are induced in various abiotic stresses including drought, salt, and ABA (Venkatesh and Park, 2015). Furthermore, DAG1, DAG2, and AtDof6 participate in seed germination (Gualberti et al., 2002; Papi et al., 2000), and CDF1, CDF2, and CDF3 are associated with the process of flowering controlled by photoperiod in Arabidopsis (Imaizumi et al., 2005). Other genes, such as AtDof2.4, AtDof5.8, and AtDof5.6/HCA2, are expressed in the early developmental stage of vascular tissue (Guo et al., 2009; Konishi and Yanagisawa, 2007). In maize, Dof1 and Dof2 participate in carbon metabolism (Yanagisawa, 2000). In rice, OsDof3 exhibits an important function in gibberellin regulation in the process of seed germination (Washio, 2003), and OsDof12 and RPBF are involved in the photoperiodic control of flowering and the regulation of seed-storage proteins, respectively (Washio, 2001; Li et al., 2009; Yamamoto et al., 2006).
Alfalfa is the most extensively cultivated leguminous forage crop and is known as the “king of herbage” (Liu et al., 2017). During its growth, alfalfa is affected by many environmental factors. Deficient water levels, low temperature and high salinity are the main limiting factors in alfalfa growth and productivity (Zhou et al., 2019). When plants suffer from these abiotic stresses, Dof TFs are responsible for improving the resistance of plants. Dof TFs have been widely reported in many species but have not been described in alfalfa because of the incomplete genomic information and scarce expression profile data for this crop (Zhou et al., 2019). However, the Noble Research Institute released the first alfalfa genome data at the diploid level in 2017, which provided important research resources for alfalfa. Therefore, to more comprehensively identify Dof TF family genes in alfalfa, a total of 36 A. thaliana, 40 M. truncatula, 37 O. sativa and 97 G. max Dof proteins were employed as queries using the cultivated alfalfa at the diploid-level genome blast server. Moreover, the expression patterns of some MsDof genes in response to ABA treatment and cold, drought, and salt stresses were analyzed by quantitative real-time PCR (qRT-PCR). The results from this study provide novel insights into the stress responses of MsDof genes and valuable information for further studies of Dof genes in alfalfa.
Materials and Methods
Identification of the Dof gene family in alfalfa
IRB approval is not needed because the research involved plants (non-transgenic plants), not humans or animals.
To more comprehensively identify the Dof TF family genes, the sequences of 36 Arabidopsis thaliana, 40 Medicago truncatula, 37 Oryza sativa, and 97 Glycine max Dof proteins were downloaded from the Plant Transcription Factor Database (Jin et al., 2016). These sequences were used as queries to search for alfalfa Dof members in the Noble Research Institute database with an E-value cutoff of 1E−5 (Altschul et al., 1990). The presence of a conserved Dof domain (PF02701) was checked based on a hidden Markov model using the Pfam Program, and the E-value was set at 1.0 (El-Gebali et al., 2019). Duplicate protein sequences of these candidate Dof family members were removed using the CD-Hit website server with the default parameters (Li and Godzik, 2006). Then, these nonredundant Dof sequences were renamed as putative MsDof genes.
The molecular weight, theoretical isoelectric point (pI), and grand average of hydropathicity (GRAVY) index value of the MsDof proteins were determined using the ProtParam Tool (Gasteiger et al., 2003). Moreover, the subcellular localization of the MsDof proteins was predicted using WoLF-PSORT (Horton et al., 2007), and orthologs of the MsDof proteins in Arabidopsis were identified by BLAST searches against Arabidopsis proteins using TAIR BLAST 2.9.0.
Functional analysis and three-dimensional structure of MsDofs
The MsDof protein functions were predicted by Gene Ontology (GO) annotation using the Blast2GO software (version 5.2) (Götz et al., 2008). First, the MsDof protein sequences were uploaded to search for similar sequences against the NCBI nonredundant database using the BLASTp tool with an e-value of 1E−3. Then, Blast2GO mapping and annotation were performed using the default parameters. Then, GO functional classification was performed on the WEGO 2.0 website, and GO enrichment analysis of the MsDof genes was conducted with agriGO v2.0, using the A. thaliana gene model (TAIR10) as a reference (Tian et al., 2017). In addition, to understand the structural properties of the MsDof proteins, the three-dimensional (3D) structures of the MsDof proteins were predicted by the PHYRE2 protein fold recognition server, and the results generated with Phyre2 were modeled with 100% confidence using the single highest scoring template (Kelley et al., 2015).
Phylogenetic analysis of MsDofs
To systemically classify the evolutionary relationships of the plant Dof gene family, a multiple-sequence alignment of the Dof protein sequences from Arabidopsis and the putative MsDof proteins was performed using ClustalW with the default parameters (Larkin et al., 2007). Then, a phylogenetic tree was constructed using MEGA (version 6.0) by the neighbor-joining method with 1000 bootstrap replicates (Tamura et al., 2013). Percentage bootstrap scores >50% are indicated at the nodes. In addition, to explore the evolutionary patterns and divergence times of the Dof genes and the selection pressure on these genes, we identified homologous pairs between alfalfa and two other grass species (A. thaliana and M. truncatula) using phylogeny-based and bidirectional best-hit methods. The nonsynonymous (Ka) and synonymous (Ks) substitution rates among alfalfa, Arabidopsis, and M. truncatula were calculated using the DnaSP 5 software, and the divergence time (T) was calculated as described in a previous study (Liu et al., 2018).
Conserved motif and gene structure analyses
To analyze the conserved Dof domain of the C2/C2-type zinc finger structure, multiple sequence alignments of these putative MsDof proteins were performed using ClustalW with the default parameters, and the sequence logos were generated using WebLogo (Crooks et al., 2004). In addition, the conserved motifs of the MsDof members were identified using MEME (version 5.1.0) (Bailey et al., 2009). The parameters of the MEME search were set as follows: the maximum number of motifs was 30; the optimum motif width was set to ≥6 and ≤200; the site distribution was set to zero or one occurrence per sequence (zoops); and the default settings were used for the other parameter options (Shu et al., 2015). Moreover, the exon-intron structures of the MsDof genes were generated on the Gene Structure Display Server using the predicted CDS and genomic sequences from the Alfalfa Breeder's Toolbox with the default parameters.
Tissue-specific expression of MsDof genes
Genome-wide transcriptome data on different alfalfa tissues were obtained from the CADL-Gene Expression Atlas provided by the Noble Research Institute (O'Rourke et al., 2015). The transcriptome data included data on six tissues: leaf, flower, pre-elonged stem, elonged stem, root, and nodule. Then, all expression data were analyzed using MEV 4.8.1 to draw a heat map of the MsDof genes.
Expression analysis of MsDof genes under abiotic stresses
Four transcriptome sequencing projects were previously performed to acquire additional genetic information on alfalfa in response to abiotic stress. These data were generated under cold (SRR7091780–SRR7091794) (Zhou et al., 2014), abscisic acid (ABA), drought, and salt treatments (SRR7160313–SRR7160357) (Luo et al., 2019a,b). In this study, we obtained the expression levels of some MsDof genes (with an absolute fold change value ≥2) through local nucleotide BLAST (BLASTN) searches against these four transcriptome datasets (Camacho et al., 2009). The expression data were analyzed using MEV 4.8.1 to perform the clustering analysis and draw the heat map.
Cis-regulatory element analysis
To investigate the potential cis-regulatory elements involved in abiotic stresses, the 2000 bp sequences upstream from the translational start sites of these stress-responsive MsDof genes were analyzed by queries in the PlantCARE database (Lescot et al., 2002). A total of eight cis-regulatory elements were identified, including ABRE (abscisic acid responsive), ARE (anaerobically induced), CGTCA-motif (methyl jasmonate responsive), DRE (dehydration, low temperature, and salt responsive), G-Box (light responsive), LTR element (low temperature responsive), TC-rich repeats (defense and stress responsive), and TCA elements (salicylic acid responsive).
Plant growth and treatments
The alfalfa seeds (Zhongmu No. 1) used in this study were provided by the Qingchuan Yang Laboratory of the Beijing Institute of Animal Sciences, Chinese Academy of Agricultural Sciences. In our experiment, all alfalfa seedlings were grown in a temperature-controlled chamber with a 16-h light/8-h dark cycle at 22°C. Before germination, the seeds were vernalized at 4°C. After 3 days of seed germination, the seedlings were transferred to a 1/2 Murashige and Skoog (MS) solution (pH = 5.8) and were watered once every 2 days. When the third alfalfa leaf appeared, these seedlings were randomly divided into five groups for different stress treatments, and six seedlings from each group were grouped separately. For the cold and heat treatments, the conditions were set as a 16-h light/8-h dark cycle at 4°C and 38°C using an artificial climate incubator, with four treatment time points (2, 6, 24, and 48 h) and one control (0 h). For the ABA treatment, the seedlings were transferred to a 1/2 MS solution containing 10 μM ABA, and four treatment time points (0, 1, 3, and 12 h) were selected. For the drought and salt treatments, the seedlings were transferred to a 1/2 MS solution containing 400 mM mannitol and 250 mM NaCl, and we designated eight treatment time points (0, 1, 3, 6, 12, and 24 h, with stress removal at 1 and 12 h). According to the method of sampling experimental materials for transcriptome sequencing described in a previous study, six whole seedlings were harvested and pooled in a freezer tube for each time point of the cold and heat treatments, and six root tips were harvested in the same way for the ABA, drought, and salt treatments. Tissues from the leaves, flowers, pre-elonged stems, elonged stems, roots, and nodules were collected from adult plants. All samples were flash-frozen in liquid nitrogen and stored at −80°C until use.
Quantitative real-time PCR analysis
The total RNA from alfalfa tissues was extracted using the TRIzol method (Sangon Biotech, Shanghai, China), according to the manufacturer's instructions. These RNAs were reverse transcribed into single-stranded cDNAs, according to the manufacturer's protocol for the FastKing RT Kit (Tiangen Biotech, Beijing, China). qRT-PCR was performed on the CFX96™ Real-Time system using a TB Green™ Premix Ex Taq™ Kit (TaKaRa, Dalian, China). Each 10 μL reaction volume contained 5 μL of a 2 SG Fast qPCR Master Mix, 1 μL of cDNA, 1 μL of a DNF buffer, 0.2 μL of each of the forward and reverse primers at 10 μM, and 2.6 μL of sterilized ddH2O. The thermal cycling conditions for PCR were as follows: denaturation at 95°C for 30 s and 40 cycles at 95°C for 5 s and at 60°C for 30 s. Each reaction was repeated three times for the different treatments, and the relative expression levels were normalized, using the Medicago actin gene as a reference gene (Liu et al., 2016). The gene expression calculation was performed as described by Pfaffl using the 2−ΔΔCq method. The primers for the MsDof genes were designed using the Primer Premier 6 software, according to the transcriptome sequence of Zhongmu No. 1. All of the primers used in this experiment are shown in Supplementary Table S1.
Results
Identification and classification of MsDof TFs
To investigate the MsDof genes in the alfalfa genome, 36 Dof protein sequences from A. thaliana, 40 from M. truncatula, 37 from O. sativa, and 97 from G. max were used to perform the BLASTp searches as queries. After verifying the presence of the Dof domain (PF02701) and obtaining nonredundant sequences, a total of 40 genes were renamed MsDof01 to MsDof40, and the protein sequences are shown in Supplementary Table S2. In addition, the MsDof family TFs were divided into eight classes (A, B1, B2, C1, C2.1, C2.2, D1, and D2) based on the analogs in the phylogenetic tree, using the previous classification of the 36 A. thaliana Dof proteins as reference (Supplementary Fig. S1). Class D1 included the largest number of MsDof TFs (30%), and Classes A and B1 accounted for 15% and 12.5% of these TFs, respectively. Three classes showed the same proportion: classes C1, C2.1, and C2.2, each showed a proportion of 7.5%. In addition, classes B2 and D2 each showed a proportion of 10%.
The physiochemical properties of these MsDof proteins were also analyzed. The lengths of 40 MsDof proteins ranged from 160 AA (MsDof28) to 1055 AA (MsDof04), with an average length of 371.35 AA, and the molecular weight ranged from 18041.33 D to 116933.39 D (Supplementary Table S3). According to the theoretical pI values, the numbers of acidic, neutral, and alkaline amino acids were 9, 6, and 25, respectively. The mean GRAVY was −0.75, and the negative GRAVY index indicated that all 40 proteins were hydrophilic (Supplementary Table S3). The subcellular localization of the MsDof proteins indicated that 39 (97.5%) proteins were nuclear localized, with the exception of MsDof38, which was chloroplast localized. Furthermore, the MsDof homologous genes in the model Arabidopsis plant were identified and are shown in Supplementary Table S3.
Functional analysis and 3D structure of MsDof proteins
The GO functional classification was analyzed and is shown in Supplementary Table S4. The GO enrichment analysis showed that 15 GO terms could be considered significantly enriched among these genes (Supplementary Table S5), and one GO term (GO:0005488) was significantly enriched for all 40 genes, indicating that most of the MsDof genes are involved in various biological processes. The 3D structures of the MsDof proteins were predicted using the PHYRE2 server to understand their structural properties. The 3D structures could be determined for only 3 of the 40 MsDof proteins, and each transmembrane helix was designated with a number indicating the residue index (Supplementary Fig. S2).
Phylogenetic analysis of MsDofs
Phylogenetic analysis of the Dof proteins from alfalfa and Arabidopsis was performed, and a circular phylogenetic tree was constructed based on the alignment of 76 amino acid sequences. These proteins were grouped into the same 9 classes included in the classification of the 36 Arabidopsis Dof proteins (Fig. 1). The Ka/Ks ratio is widely used to measure genetic evolution and selection pressure. Scatter plot statistics showed that the ratios of two homologous pairs (MsDof03/MsDof07 and MsDof20/MsDof25) were greater than 1, indicating that they were subjected to positive selection during evolution, while the others were shown to be subjected to negative or stabilizing selection (Fig. 2). In addition, the divergence times between 10 paralogous pairs showed that the MsDofs underwent duplication events from between ∼0.59 and 60.73 million years ago (MYA). Also, the analysis of 29 orthologous pairs showed that the MsDofs and MtDofs were separated ∼2.48 to 12.05 MYA (Supplementary Table S6).

Phylogenetic relationships of alfalfa and Arabidopsis Dof gene families. The unrooted phylogenetic tree was constructed using MEGA 6.0 and the neighbor-joining method. The bootstrap test was performed with 1000 replications. The black circle indicates the Dof genes in alfalfa, and the gray circle indicates the Dof genes in Arabidopsis. The different branches indicate the different subfamilies.

Scatter plot statistics of the Ka and Ks values for grass species. The black dotted line with a slope of one is used to indicate Ka/Ks = 1. The black, gray and light gray circles indicate homologous pairs of different types. Ms-At, orthologous pairs of Dofs between Medicago sativa and Arabidopsis thaliana; Ms-Ms, paralogous pairs of MsDofs; Ms-Mt, orthologous pairs of Dofs between M. sativa and Medicago truncatula.
Conserved Dof domains and gene structure
Multiple sequence alignments of these 40 MsDof proteins were performed using ClustalW to investigate the classic sequence characteristics of C2/C2-type zinc fingers. In general, the basic regions of the Dof domain consist of ∼50–52 amino acid residues located in the N-terminal region. As expected, a highly conserved Dof domain was identified in all MsDof proteins (Supplementary Fig. S3). Then, the WebLogo program was applied to explore the frequency of most prevalent amino acids at each position within the alfalfa Dof domain (Fig. 3). The bit value indicates the information content at each position in the sequence. In addition, a total of 30 conserved motifs were found among all 40 MsDofs using MEME, and the amino acid consensus sequence of each motif is listed in Supplementary Table S7. The results showed that the widths of the 30 motifs ranged from 9 to 200 AA, and the E-values ranged from 5.8E−98 to 1.7E+00 (Supplementary Table S7). As shown in Supplementary Figure S1, most of the closely related MsDofs in the phylogenetic tree presented a similar distribution of conserved motifs, and every MsDof protein contained motif1, which provides powerful support for the results of the phylogenetic analysis.

Dof domains are highly conserved across all Dof proteins in alfalfa. The sequence logos are based on alignments of all alfalfa Dof domains. Multiple alignment analysis of 40 typical alfalfa Dof domains was performed using ClustalW. The bit score indicates the information content for each position in the sequence. The triangles indicate the conserved cysteine residues (Cys) in the Dof domains, and the asterisks indicate the conserved residues that are identical among all MsDof domains.
Furthermore, to explore the exon-intron structure characteristics, all 40 putative MsDof genes were analyzed using the Gene Structure Display Server. As shown in Supplementary Figure S4, the predicted number of exons varied from one to eight, and the number of introns varied from zero to seven. Twelve (30%) of the MsDof genes were intronless, whereas 23 (57.5%) contained 1 intron, 3 (7.5%) contained 3 introns, MsDof04 had 4 introns, and MsDof08 had 7 introns. These similar structural characteristics may be related to the functions of these genes in the alfalfa genome.
Expression profile analysis of the MsDof genes in alfalfa tissues
Microarray data for the alfalfa B47 genotype were downloaded from the CADL Gene Expression Atlas and used to assess the transcript abundance profiles of the MsDof genes in six tissues (leaf, flower, pre-elonged stem, elonged stem, root, and nodule). Only 33 MsDof genes were included in the analysis of the transcript abundance, because the others were not found in the CADL Gene Expression Atlas database. To visualize the expression data, an expression heat map was constructed using the program, MEV 4.8.1. The 33 MsDof genes were divided into 6 subgroups (A to F) according to their expression patterns. As shown in Figure 4, subgroup A contained 8 genes that showed the highest transcript accumulation levels in the flower tissue, and 3 of the 33 MsDof genes (subgroup B) showed the highest transcript accumulation levels in the leaf tissue. Subgroup C included five MsDof genes that showed a lower transcript accumulation in the roots or nodules. By contrast, some of these genes showed higher transcript accumulation levels in the leaf, flower, pre-elonged stem, or elonged stem tissue. Moreover, five MsDof genes belonging to subgroup D were highly expressed in the flowers, elonged stems, or roots and weakly expressed in the other three tissues. In addition, 5 of the 33 MsDof genes (subgroup E) showed higher transcript accumulation levels in the pre-elonged stems, roots, or nodules, and the remaining 7, belonging to subgroup F, showed a higher transcript accumulation in pre-elonged stems or elonged stems and lower levels in leaf or nodule tissues.
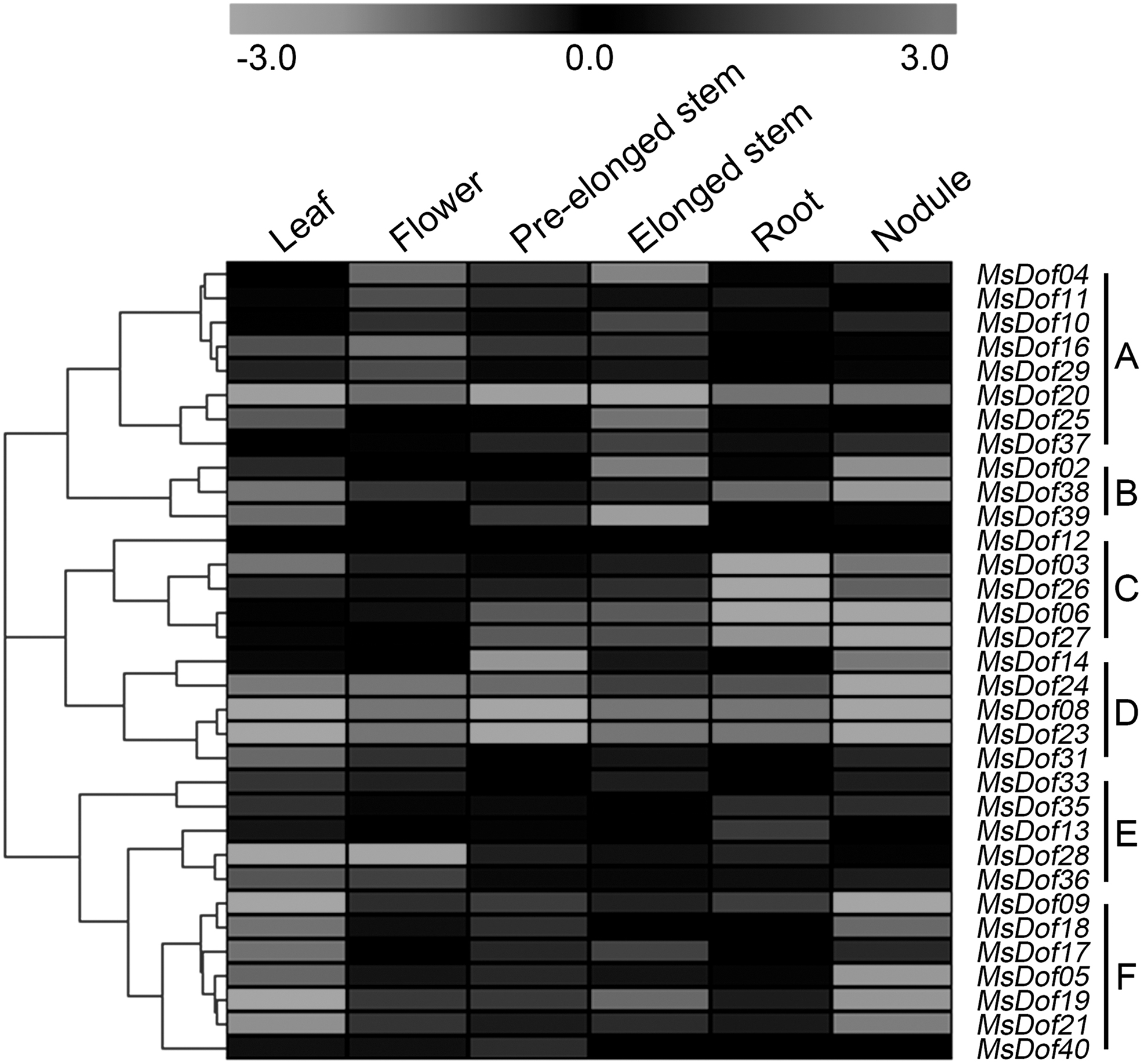
Heat map representation of the expression profiles of the MsDofs among different tissues. The heat map was generated using MEV 4.8.1. Higher and lower levels of transcript accumulation are indicated in gray, dark gray, and light gray, respectively, and the median level is indicated in black. Different subgroups indicate various transcription levels in different tissues.
Expression analysis of MsDof genes in response to abiotic stresses
The expression levels of the MsDof genes under abiotic stresses were determined through BLASTN searches against our four transcriptome datasets. As a result, seven, six, seven, and seven different MsDof genes were identified under cold, ABA, drought, and salt stresses, respectively. As shown in Figure 5, the seven MsDof genes identified under cold stress were divided into two groups, and MsDof10 and MsDof35 (subgroup A) were induced later than the other genes, which were induced in the early stage, compared to the control. Under ABA treatment, subgroup A was inhibited in the late stage, thus differing from subgroup B. As shown in Figure 6, MsDof14 was induced by drought and salt stresses earlier than the other genes, and MsDof01, MsDof02, MsDof10, MsDof35, and MsDof39 were induced later than the other genes under both the drought and salt treatments.
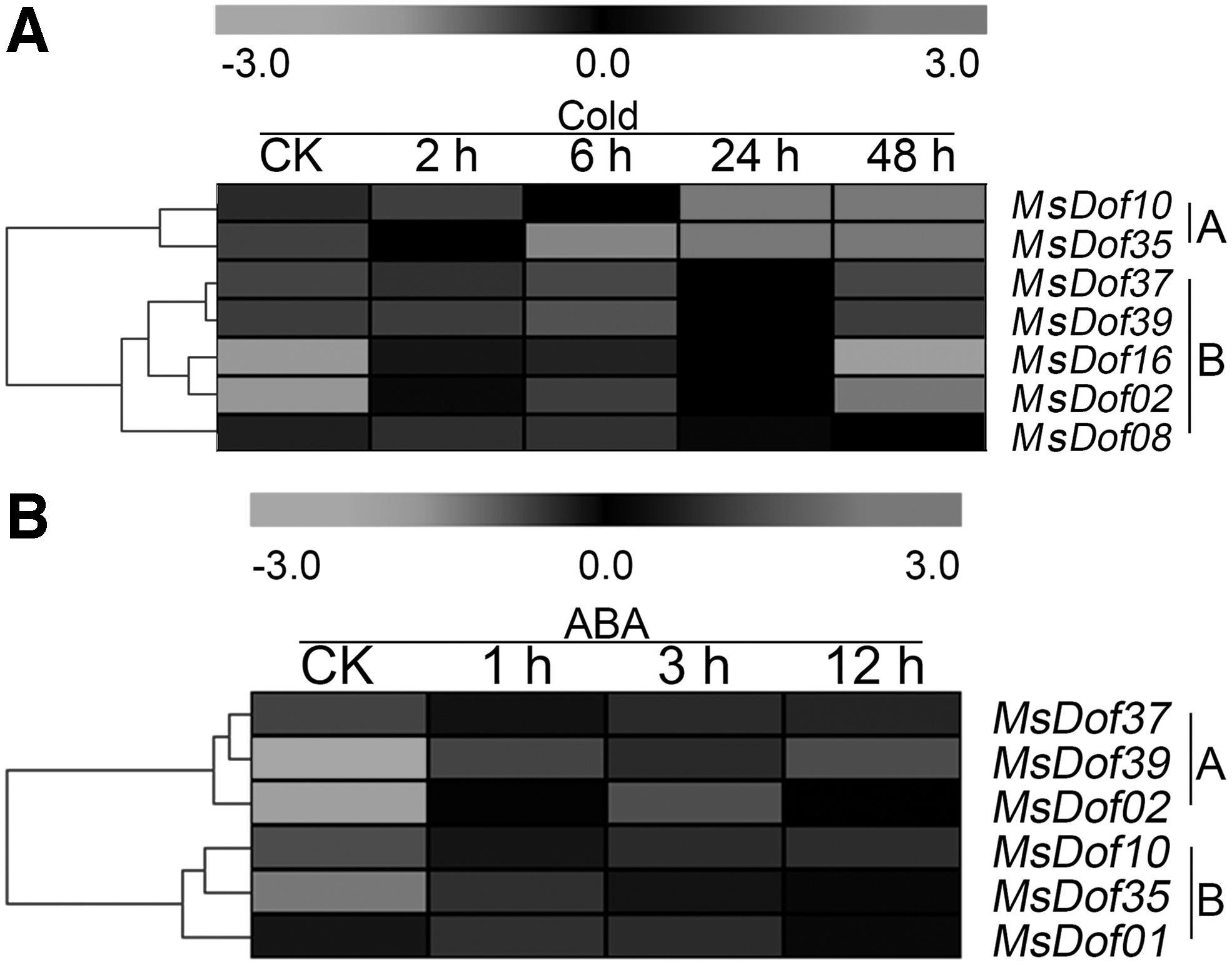
Expression of MsDof genes in response to cold and ABA treatments. The heat map was generated using MEV 4.8.1. It shows the changes in the expression levels of these MsDof genes at different time points after treatment
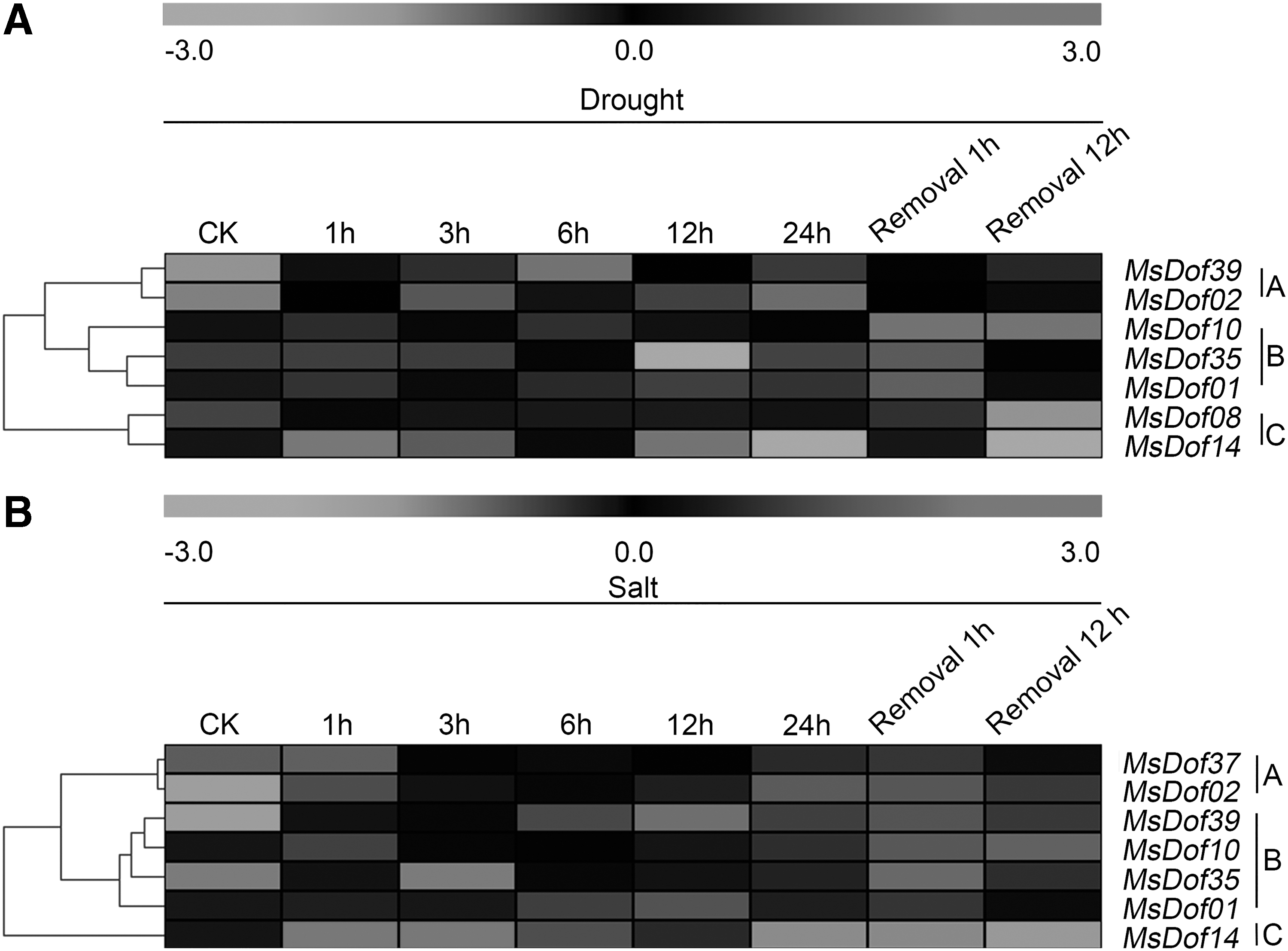
Expression of MsDof genes in response to drought and salt treatments. The heat map was generated using MEV 4.8.1. It shows the changes in the expression levels of these MsDof genes at different time points after treatment with
Cis-regulatory elements in MsDof gene promoters
Cis-regulatory elements located in promoter regions are related to the expression patterns of abiotic stress-responsive genes. Thus, we explored the distribution of eight elements in nine MsDof stress-responsive gene promoters. The results showed that all nine MsDof genes containing ABRE and G-Box exhibited binding to ABA-responsive and light-responsive elements. In addition, we found three MsDof genes (MsDof08, MsDof35, and MsDof10) containing LTR elements, among which only MsDof10 contained a DRE that was responsive to dehydration, low temperature, and salt stresses (Fig. 7).
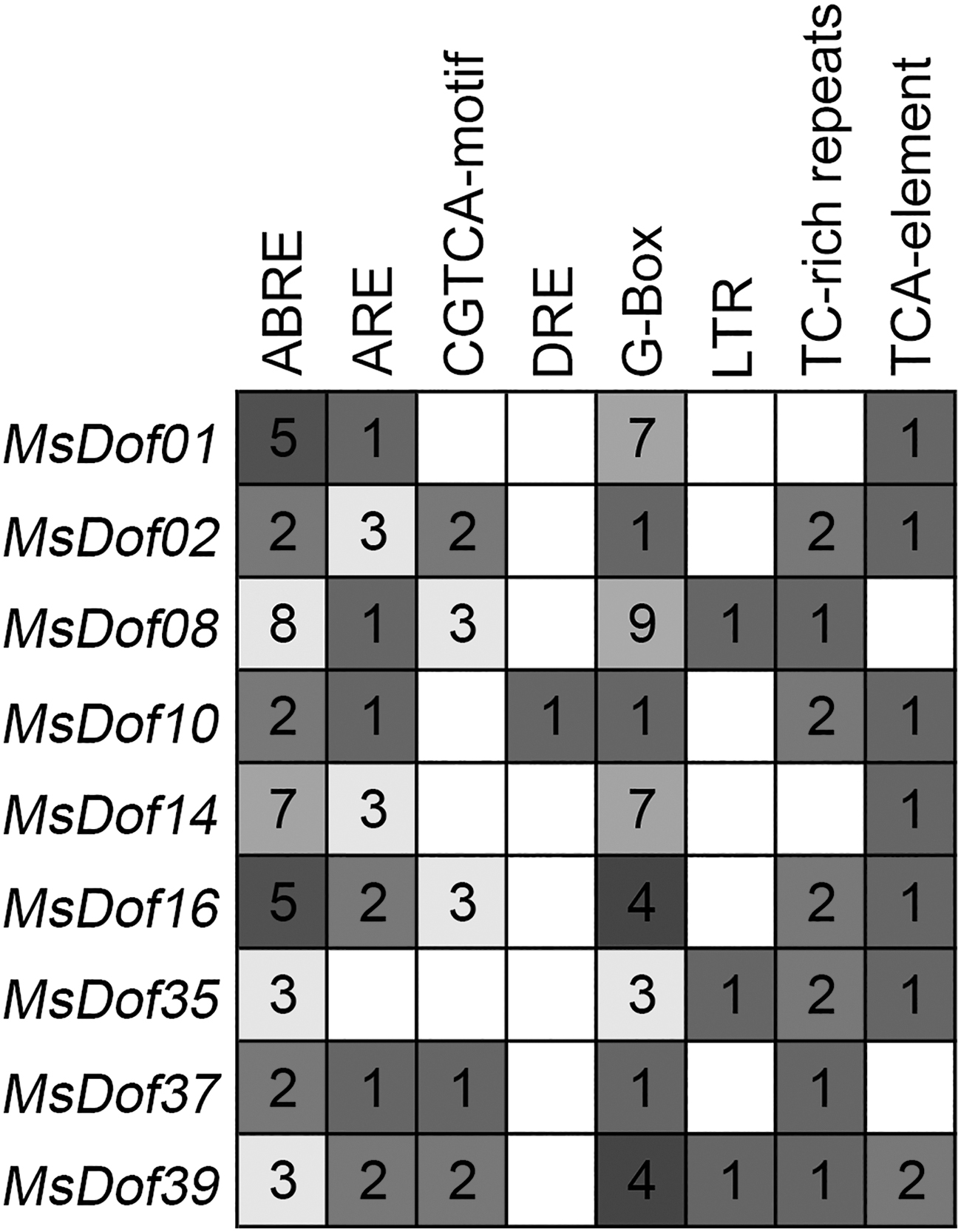
Cis-regulatory elements in the promoter regions of MsDofs. The different shaded blocks with a number represents the number of cis-elements in the analyzed promoter region of the indicated MsDofs. The sequences of the regions 2000 bp from the promoters of nine MsDof genes were downloaded from the alfalfa databases provided by the Noble Research Institute.
Gene expression analysis qRT-PCR validation
To verify the expression profiles in different tissues of MsDof genes, nine tissue-specific expression genes were selected from Figure 4, and their expression levels were analyzed using qRT-PCR. The results reveal spatial variations in the expression of MsDof genes in different alfalfa organs, and all genes were expressed in distinct patterns (Fig. 8). Genes such as MsDof03, MsDof26, and MsDof39 were highly expressed in the leaves, and the expression levels of MsDof02, MsDof03, MsDof09, MsDof16, MsDof26, MsDof37, and MsDof39 were relatively higher in flowers. Interestingly, the expression levels of MsDof02, MsDof03, MsDof06, MsDof17, and MsDof39 showed that all the five genes were more highly expressed in elonged stems than in pre-elonged stems. Meanwhile, all the nine genes had a relatively lower expression in roots, and only MsDof02 and MsDof39 were more highly expressed in nodules.
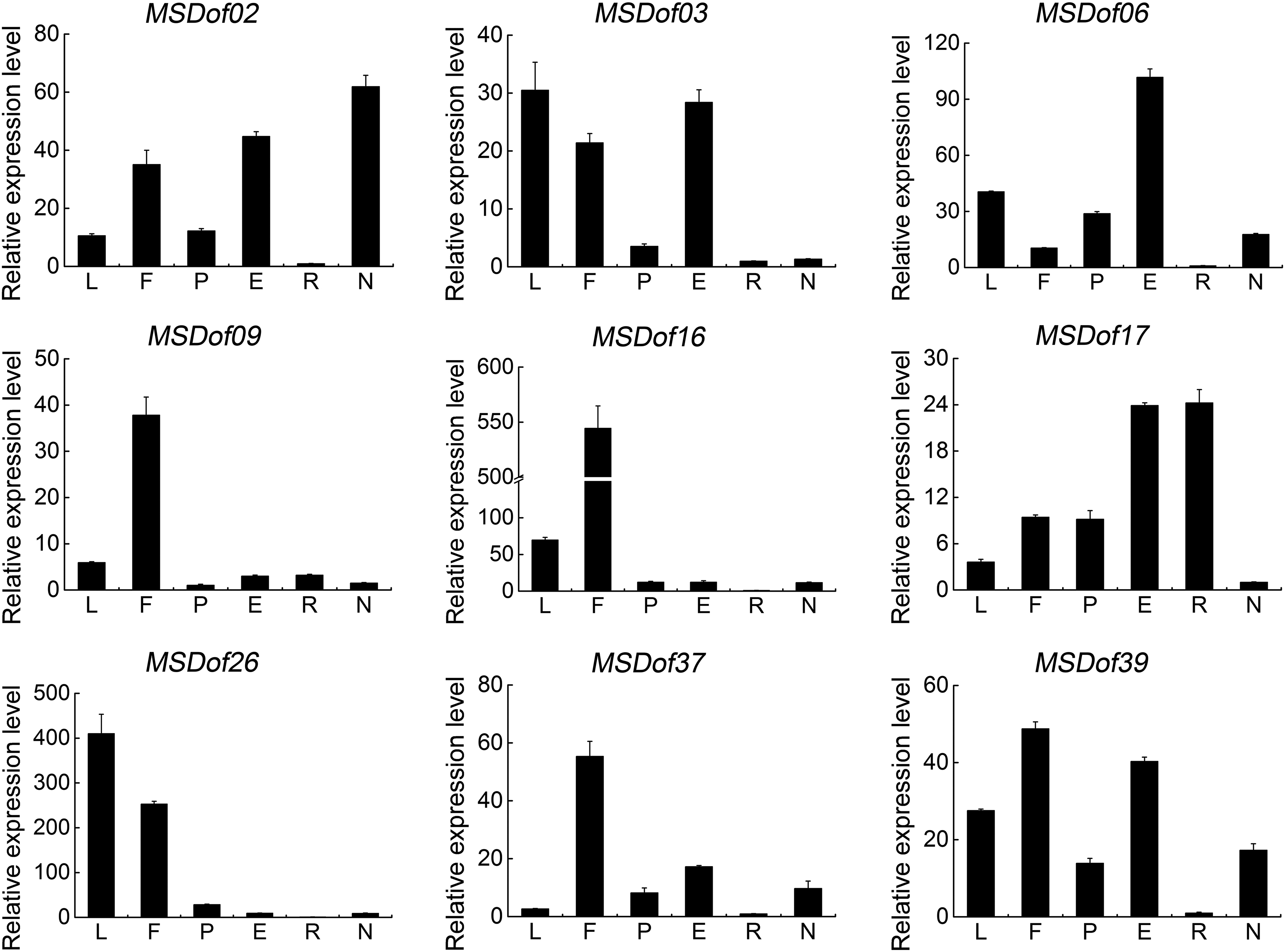
Expression profiles of MsDof genes in various tissues. qRT-PCR analyses were used to assess the MsDof transcript levels in L: leaf, F: flower, P: pre-elonged stem, E: elonged stem, R: root, and N: nodule. The error bars were obtained from three measurements. qRT-PCR, quantitative real-time PCR.
The functions of MsDof genes in response to abiotic stresses are unknown. To confirm the roles of MsDof genes in response to abiotic stresses, we selected the significantly induced MsDof genes and analyzed their expression profiles under abiotic stresses using qRT-PCR. As a result, MsDof10, MsDof35, and MsDof39 were found to be significantly induced under all five treatments. Moreover, the results showed that all the selected genes were induced to varying degrees by cold (Fig. 9), heat (Fig. 10), ABA (Fig. 11), drought (Fig. 12), and salt (Fig. 13) stresses.

Expression profiles of MsDof genes in response to cold stress treatment. qRT-PCR analyses were used to assess the MsDof transcript levels in whole seedlings sampled at 0, 2, 6, 24, and 48 h after exposure to the 4°C treatment. The error bars were obtained from three measurements. Lowercase letters above the bars indicate significant differences (p < 0.05) among the treatments.
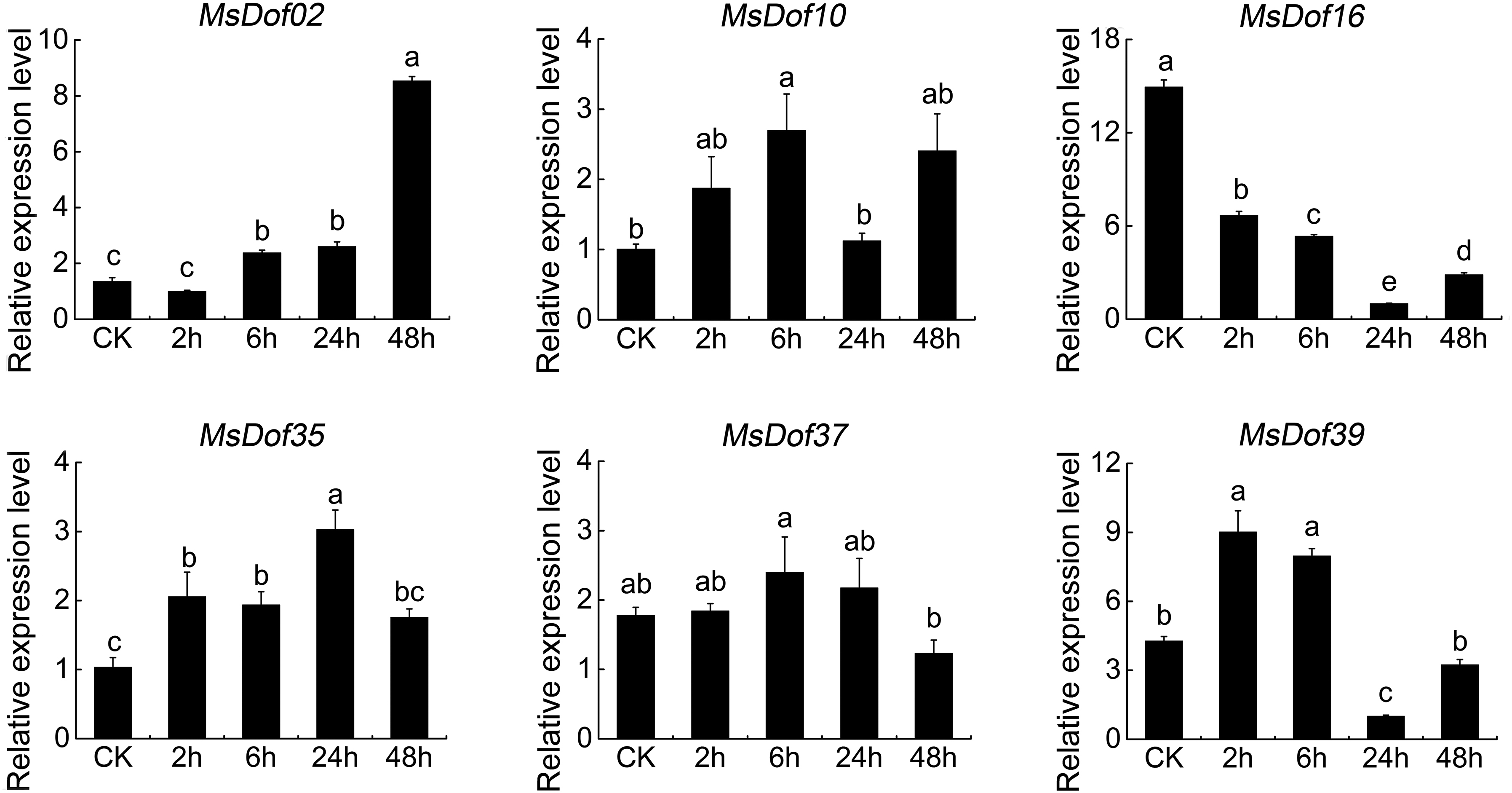
Expression profiles of MsDof genes in response to the heat stress treatment. qRT-PCR analyses were used to assess the MsDof transcript levels in whole seedlings sampled at 0, 2, 6, 24, and 48 h after exposure to the 38°C treatment. The error bars were obtained from three measurements. Lowercase letters above the bars indicate significant differences (p < 0.05) among the treatments.
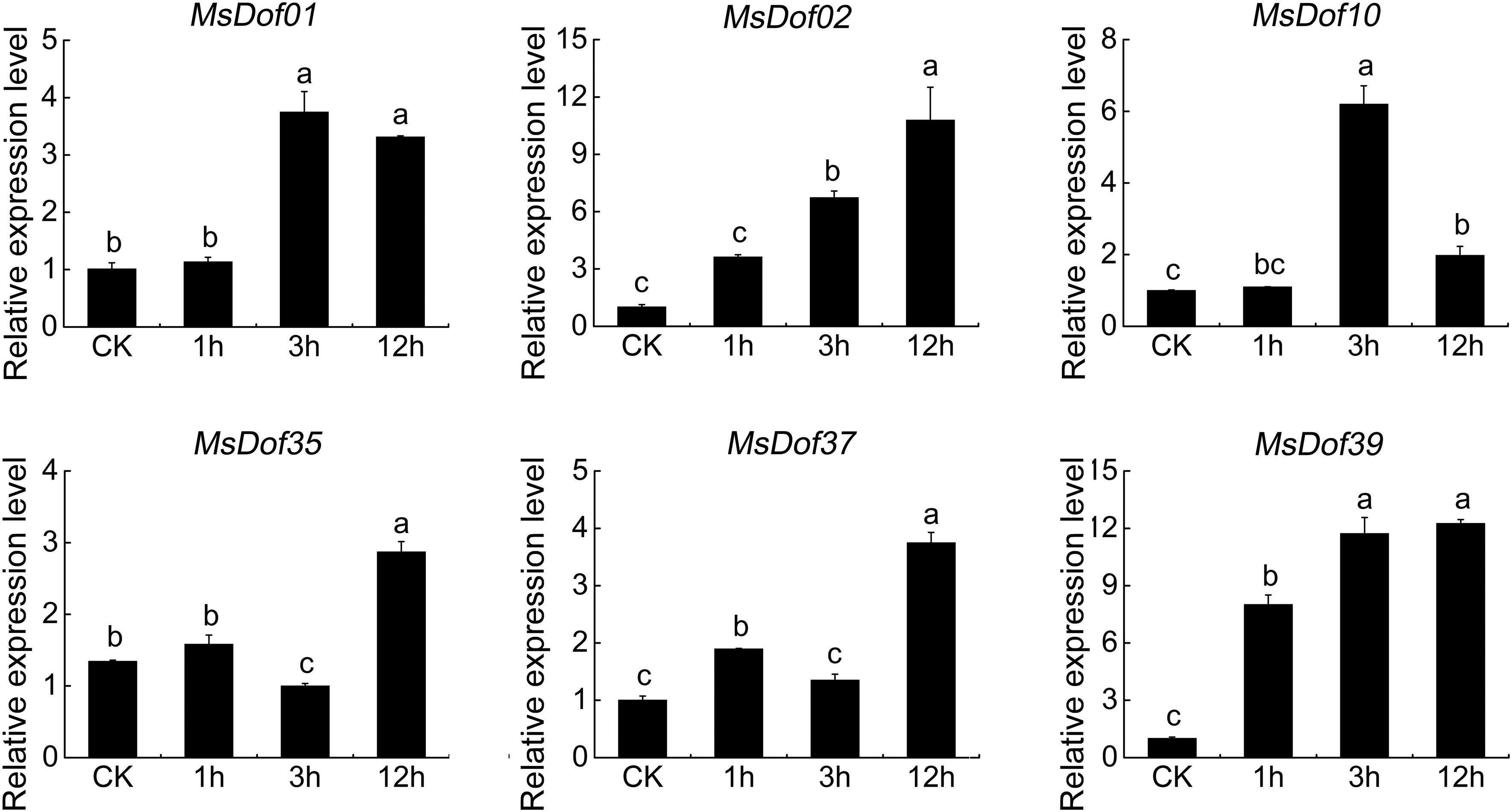
Expression profiles of MsDof genes in response to the ABA treatment. qRT-PCR analyses were used to assess the MsDof transcript levels in the root tips sampled at 0, 1, 3, and 12 h after exposure to the 10 mM abscisic acid treatment. The error bars were obtained from three measurements. Lowercase letters above the bars indicate significant differences (p < 0.05) among the treatments.
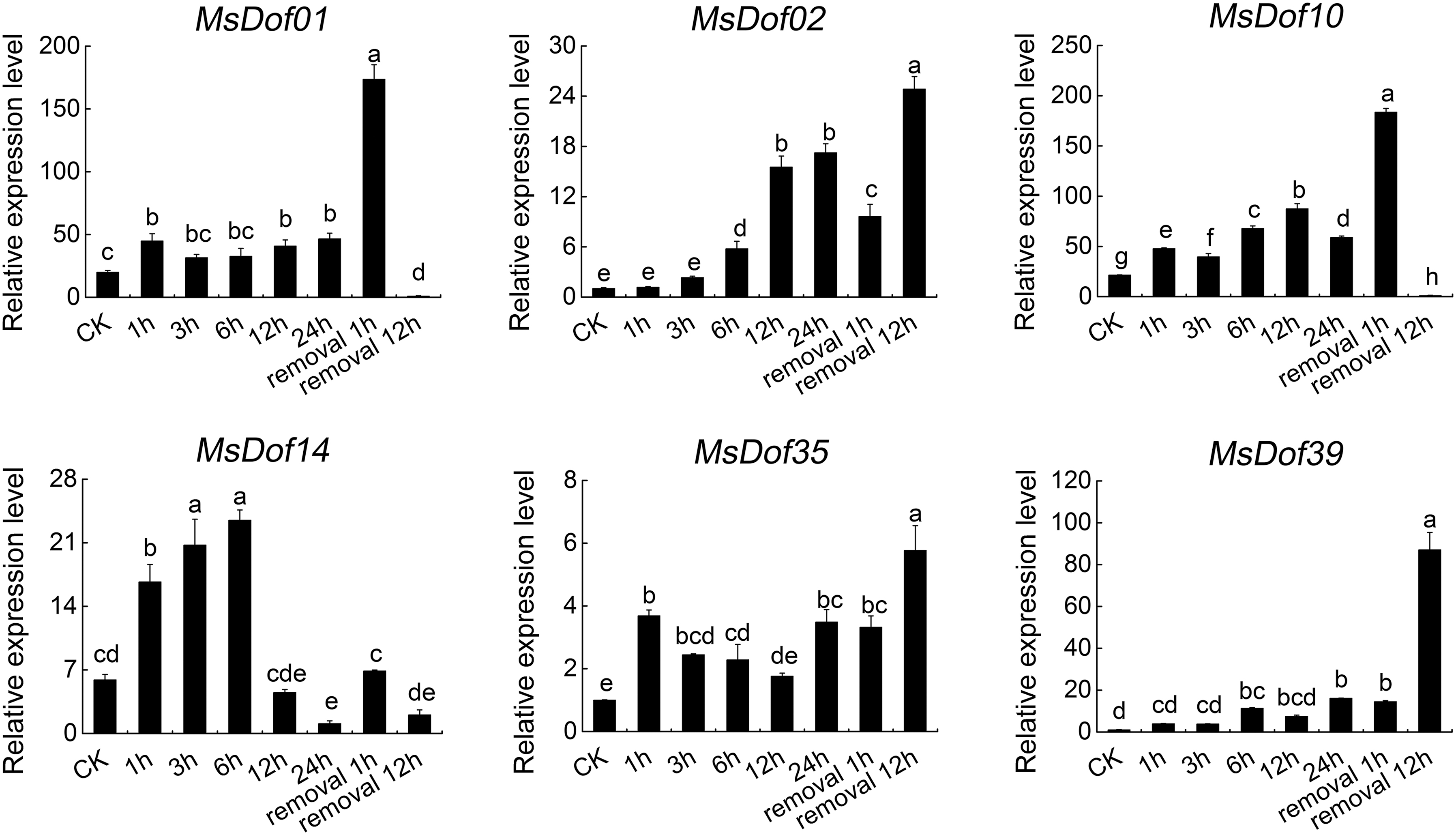
Expression profiles of MsDof genes in response to the drought treatment. qRT-PCR analyses were used to assess the MsDof transcript levels in the root tips sampled at 0, 1, 3, 6, 12, and 24 h, with removal at 1 and 12 h, after exposure to the 400 mM mannitol treatment. The error bars were obtained from three measurements. Lowercase letters above the bars indicate significant differences (p < 0.05) among the treatments.

Expression profiles of MsDof genes in response to the salt treatment. qRT-PCR analyses were used to assess the MsDof transcript levels in the root tips sampled at 0, 1, 3, 6, 12 h, and 24 h, with removal at 1 and 12 h, after exposure to the 250 mM NaCl treatment. The error bars were obtained from three measurements. Lowercase letters above the bars indicate significant differences (p < 0.05) among the treatments.
Discussion
Gene family analysis has become an important approach for analyzing gene function, evolution, and structure. The Dof family of plant-specific transcription factors was first isolated in maize and shown to be involved in C4 photosynthesis (Yanagisawa, 2002). Several studies have addressed the genome-wide identification, functions, and evolutionary relationships of Dofs in many species whose genomes have been sequenced. These species include M. truncatula (Shu et al., 2015), Arabidopsis, rice (Lijavetzky et al., 2003), sorghum (Kushwaha et al., 2011), pigeon pea (Malviya et al., 2015), potato (Venkatesh and Park, 2015), Chinese cabbage (Ma et al., 2015), pepper (Kang et al., 2016), carrot (Huang et al., 2016), tomato (Cai et al., 2013), cucumber (Wen et al., 2016), poplar (Yang et al., 2006), wheat (Shaw et al., 2009), and soybean (Guo and Qiu, 2016). Interestingly, we found that the number of reported Dof members shows little diversity among different plant species, regardless of their genome size. In a previous study, Dof transcription factors were found to exhibit significant functions in many biological and physiological processes regulating growth, development, and defense mechanisms and flowering in plants. For example, BPBF (HvDof24), SAD (HvDof23), HvDof17, and HvDof19 were reported to be involved in the regulation of hormonal balance between gibberellin and abscisic acid during seed germination (Diaz et al., 2002, 2005; Moreno-Risueno et al., 2007). Nevertheless, little is known about these genes in alfalfa. In our study, we conducted a comparative analysis of the Dof family between alfalfa and Arabidopsis to analyze the evolutionary relationships of these genes and determine their potential functions in response to abiotic stresses. We identified 40 putative MsDof genes in the alfalfa genome. To clarify the phylogenetic relationships of the MsDof gene family, a combined phylogenetic tree was constructed based on the alignment of Arabidopsis and alfalfa amino acid sequences. Consistent with a previous study (Lijavetzky et al., 2003), the 76 MsDof genes were classified into 4 groups (A to D) and 9 subgroups (A, B1, B2, C1, C2.1, C2.2, C3, D1, and D2) based on the neighbor-joining phylogenetic tree (Fig. 1), but in alfalfa, all 40 MsDof genes were divided into 4 groups and 8 subgroups (A, B1, B2, C1, C2.1, C2.2, D1, and D2) (Supplementary Fig. S1). No MsDof gene in class C3 was found, which is similar to previous reports (Li et al., 2016; Wen et al., 2016). As found in Arabidopsis, class D1 was the largest subgroup in alfalfa. In addition, the motif of MsDofs was highly conserved and most of the closely related MsDof genes in the phylogenetic tree presented a similar amount of exon and intron. These results suggest that the predicted MsDofs identified in our research can be used as valuable resources for the functional analysis of MsDofs.
We calculated the divergence times (T) of paralogous pairs and orthologous pairs in the alfalfa Dof family using Ks values. The divergence time of monocots/dicots has been estimated to be ∼170 to 235 MYA (Blanc and Wolfe, 2004). The Arabidopsis/tomato and pepper/tomato divergence times have been predicted to be at ∼110 to 130 MYA and 19.2 to 20 MYA, respectively (Wang et al., 2008; Kim et al., 2014). In this study, we predicted that the duplications occurred in 10 paralogous Dof pairs in alfalfa and the 29 orthologous Dof pairs between alfalfa and M. truncatula. The duplication of most of the MsDofs clearly occurred before the MsDof/MtDof divergence, whereas only three paralogous pairs (MsDof03/MsDof07, MsDof06/MsDof27, and MsDof15/MsDof26) were duplicated recently. The Ka/Ks ratio provides a measure of genetic evolution and selection pressure. If the ratio is greater than 1, it indicates positive selection, while a ratio equal to 1 indicates neutral selection, and a ratio less than 1 indicates negative or stabilizing selection (Liu et al., 2018). In this study, the Ka/Ks ratios of two homologous pairs were >1, suggesting that the duplicated pairs evolved under positive selection, while others were indicated to be under negative selection or stabilizing selection. The Ka/Ks ratios of all duplicated MsDof/MtDof pairs were <1, indicating that the duplicated pairs evolved under negative selection. The duplication analysis indicates that the segmental expansion of the MsDofs might have occurred before the alfalfa and M. truncatula separation event, and after the duplications, the functional divergence of the MsDofs was retained.
Previous studies have explored the association of Dof genes with plant tissue-specific expression and organ development (Gupta et al., 2015; Venkatesh and Park, 2015). Vascular tissue is abundant in plant stems and roots. In Arabidopsis, AtDof2.4 and AtDof5.8 have been proposed to function in the early stage, but to be involved in different processes related to vascular development (Konishi and Yanagisawa, 2007; Gardiner et al., 2010). Dof5.6/HCA2 induces the formation of the interfascicular cambium and regulates vascular tissue development (Guo et al., 2009). In this study, MsDof09 and MsDof19, exhibiting a maximum similarity to AtDof5.8 (At5g66940) and AtDof5.6 (At5g62940), respectively, in evolutionary relationship, showed relatively high transcript abundance profiles in elonged stems and roots, according to the heat map. Thus, we predicted that MsDof09 and MsDof19 would exhibit similar expression patterns to AtDof5.8 and AtDof5.6, respectively. AtOBP3 (At3g55370), which regulates phytochrome and cryptochrome signaling (Ward et al., 2005), showed a high similarity to MsDof31, indicating similar expression patterns. The expression of MsDof16 was significantly higher in flowers than in other organs (Figs. 4 and 8), indicating that MsDof16 plays more important roles in reproductive organs than in vegetative organs. These expression patterns were similar to those of their Arabidopsis homolog, AtCDF2 (At5g39660), which regulates the timing of the transition from the vegetative to the reproductive phase (Fornara et al., 2009). In addition, MsDof03, MsDof38, and MsDof39 showed the highest expression levels in leaves, whereas MsDof20 was exhibited in nodules, indicating a different role in alfalfa development. Meanwhile, all the nine genes verified using qRT-PCR showed a lower expression level in pre-elonged stems and a higher expression level in elonged stems, indicating that these genes are mainly expressed later in plant development, which implies that different genes have a different expression pattern in distinct development stages (Fig. 8). No research has shown that MsDof genes directly regulate plant development, but tissue-specific analysis suggests that some of these genes may be involved in biological functions during plant development.
Dof genes have been shown to be involved in abiotic stress responses among many plant species (Corrales et al., 2014; Gupta et al., 2015), but in alfalfa, their exact roles in abiotic tolerance are not known. Understanding MsDof functions in alfalfa will help to characterize the cold, heat, drought, and salt tolerance of this crop. In previous studies, the roles of the Dof genes under abiotic stresses were investigated using various transgenic plants. For example, the overexpression of the Arabidopsis Dof gene, CDF3, was found to enhance resistance to drought, cold, and osmotic stress in transgenic A. Thaliana (Corrales et al., 2017). Similarly, transgenic A. thaliana overexpressing SlCDF1 or SlCDF3 (tomato Dof factor) was shown to exhibit a lower sensitivity to drought and salt stresses, compared to the wild type (Corrales et al., 2014). The expression profiles of many Dof genes indicate that their functions may be involved in responses to abiotic stresses. For example, OBP2 (AtDof1.1) is markedly induced after MeJA treatment and mechanical injury in A. thaliana (Skirycz et al., 2006), and most of the BraDof genes are rapidly induced by cold, heat, salt, and drought treatments in Chinese cabbage (Ma et al., 2015). In this study, the expression profiles of some MsDof genes exposed to ABA, cold, heat, drought, and salt were investigated using qRT-PCR in alfalfa seedlings and supported by transcriptome sequencing data. All of these MsDof genes were shown to be significantly induced by cold, heat, ABA, drought, or salt (Figs. 9–13). Most of the MsDof genes were upregulated quickly by cold, heat, and salt treatments, although a notable exception may be observed: some genes were downregulated quickly under ABA and drought treatments. These results may be due to the different mechanisms of the MsDof gene response under different treatments, and different MsDof genes may also affect each other. However, the degree of the observed expression fold changes varied between the RNA-seq and qRT-PCR experiments. This phenomenon has also appeared in previous studies (Wang et al., 2015; Dong et al., 2017; Cao et al., 2018; Zhang et al., 2018). It may be due to differences in the sample collection or genetic diversity between individual alfalfa plants. In this study, MsDof10, MsDof35, and MsDof39 were highly induced under ABA, cold, heat, drought, and salt treatments. Therefore, these genes can be used as candidate genes that may participate in abiotic stress responses. Cis-regulatory elements that act as binding sites for TFs are generally related to the expression patterns of abiotic stress-responsive genes. In fact, several common putative cis-regulatory elements have been confirmed in upstream regions of MsDof abiotic stress-responsive genes, such as DRE-, LTR-, and TC-rich repeats, and we found that all the genes contained ABRE and G-Box elements (Fig. 7). Moreover, a previous study showed that ABRE is the major type of cis-element related to ABA-responsive gene expression (Yamaguchi-Shinozaki and Shinozaki, 2006). These results demonstrate the potential positive functions of MsDof genes in plant adaptation to abiotic stresses; however, further study is needed to confirm their roles.
Conclusion
In this study, we performed a comprehensive analysis of the Dof genes in alfalfa. A total of 40 genes were identified in the alfalfa genome, and these MsDof TFs were divided into eight classes: A, B1, B2, C1, C2.1, C2.2, D1, and D2. The analysis of the physiochemical properties, phylogenetic relationships, and structures of the genes indicated that they were mostly similar within the same class. The analysis of the duplications between alfalfa and M. truncatula showed that the segmental expansion of the MsDofs might have occurred before the separation of these two species. Moreover, the tissue expression patterns demonstrated that most of the MsDof genes were expressed in specific tissues. In addition, we identified 8 candidate genes that may participate in abiotic stress responses, and we confirmed their expression profiles by qRT-PCR. Our study aids in understanding the roles of MsDof genes under abiotic stresses and provides resources for further investigations.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
The research was supported by the National Natural Science Foundation of China, grant/award numbers: 31722055 and 31672476.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Table S7
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.